 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeContextualizing Biological Language Models across Modalities via Logit-Space Contrastive Alignment
Jun 17, 2026Pretrained biological language models expose per-token probability distributions through masked-token prediction, providing the likelihood interface central to sequence design, variant scoring, and mechanistic interpretation. Yet these distributions are learned from broad unlabeled corpora and are not naturally conditioned on task-specific biological contexts such as interaction partners, cellular environments, or therapeutic interventions. Existing contextual matching methods often distort this interface through pooled embeddings, contrastive latent spaces, or task-specific prediction heads. We introduce LOGICA (Logit-space Contrastive Alignment), a framework for context-conditioned prediction that performs contrastive learning directly in output-logit space. Using gated cross-modal adapters compatible with each model's native token head, LOGICA preserves the pretrained likelihood interface and converts contextualized token log-likelihoods into matching scores. Alignment is defined through context-sensitive token probabilities rather than proximity in a shared embedding space, enabling learning from sparse paired data across models with distinct vocabularies, without a shared tokenizer or decoder. LOGICA is particularly effective for mutation-local variant ranking, where comparisons reduce to context-conditioned likelihoods of mutant tokens at perturbed sites. Across protein--ligand binding, TCR--peptide activity, and drug-conditioned resistance prediction, LOGICA improves over prior state-of-the-art methods, including matched latent-contrastive and conditional MLM baselines, while retaining a token-level interface for interpretation and generation. On held-out-gene single-mutation drug-resistance prediction, LOGICA improves AUC from near-random latent-space baselines of $\sim$0.55 to $\sim$0.65.
Feature selection as Monte-Carlo Search in Growing Single Rooted Directed Acyclic Graph by Best Leaf Identification
Nov 19, 2018
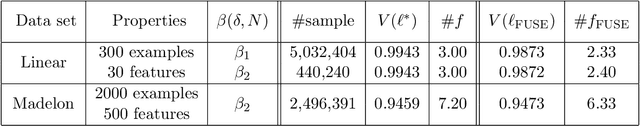
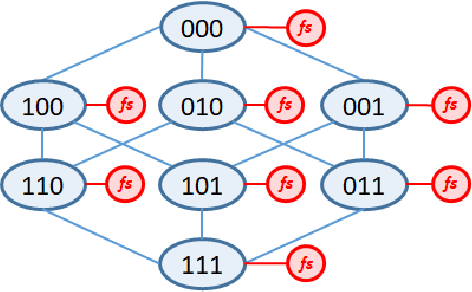
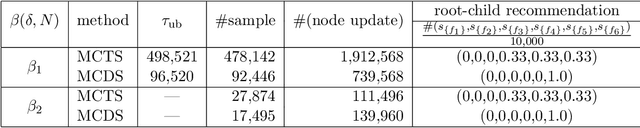
Monte Carlo tree search (MCTS) has received considerable interest due to its spectacular success in the difficult problem of computer Go and also proved beneficial in a range of other domains. A major issue that has received little attention in the MCTS literature is the fact that, in most games, different actions can lead to the same state, that may lead to a high degree of redundancy in tree representation and unnecessary additional computational cost. We extend MCTS to single rooted directed acyclic graph (SR-DAG), and consider the Best Arm Identification (BAI) and the Best Leaf Identification (BLI) problem of an expanding SR-DAG of arbitrary depth. We propose algorithms that are (epsilon, delta)-correct in the fixed confidence setting, and prove an asymptotic upper bounds of sample complexity for our BAI algorithm. As a major application for our BLI algorithm, a novel approach for Feature Selection is proposed by representing the feature set space as a SR-DAG and repeatedly evaluating feature subsets until a candidate for the best leaf is returned, a proof of concept is shown on benchmark data sets.