 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerated Hydration Site Localization and Thermodynamic Profiling
Nov 23, 2024



Water plays a fundamental role in the structure and function of proteins and other biomolecules. The thermodynamic profile of water molecules surrounding a protein are critical for ligand binding and recognition. Therefore, identifying the location and thermodynamic behavior of relevant water molecules is important for generating and optimizing lead compounds for affinity and selectivity to a given target. Computational methods have been developed to identify these hydration sites, but are largely limited to simplified models that fail to capture multi-body interactions, or dynamics-based methods that rely on extensive sampling. Here we present a method for fast and accurate localization and thermodynamic profiling of hydration sites for protein structures. The method is based on a geometric deep neural network trained on a large, novel dataset of explicit water molecular dynamics simulations. We confirm the accuracy and robustness of our model on experimental data and demonstrate it's utility on several case studies.
Accurate Free Energy Estimations of Molecular Systems Via Flow-based Targeted Free Energy Perturbation
Feb 23, 2023The Targeted Free Energy Perturbation (TFEP) method aims to overcome the time-consuming and computer-intensive stratification process of standard methods for estimating the free energy difference between two states. To achieve this, TFEP uses a mapping function between the high-dimensional probability densities of these states. The bijectivity and invertibility of normalizing flow neural networks fulfill the requirements for serving as such a mapping function. Despite its theoretical potential for free energy calculations, TFEP has not yet been adopted in practice due to challenges in entropy correction, limitations in energy-based training, and mode collapse when learning density functions of larger systems with a high number of degrees of freedom. In this study, we expand flow-based TFEP to systems with variable number of atoms in the two states of consideration by exploring the theoretical basis of entropic contributions of dummy atoms, and validate our reasoning with analytical derivations for a model system containing coupled particles. We also extend the TFEP framework to handle systems of hybrid topology, propose auxiliary additions to improve the TFEP architecture, and demonstrate accurate predictions of relative free energy differences for large molecular systems. Our results provide the first practical application of the fast and accurate deep learning-based TFEP method for biomolecules and introduce it as a viable free energy estimation method within the context of drug design.
On-the-fly Prediction of Protein Hydration Densities and Free Energies using Deep Learning
Jan 07, 2020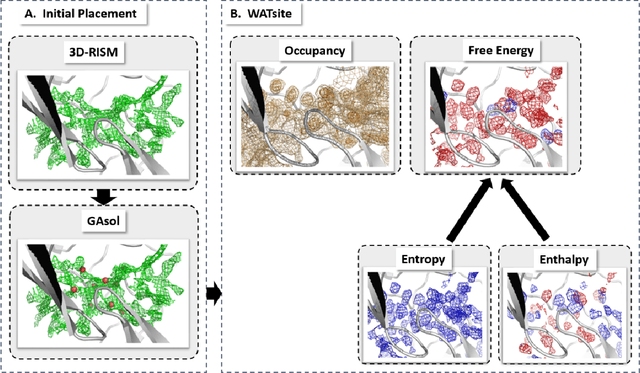
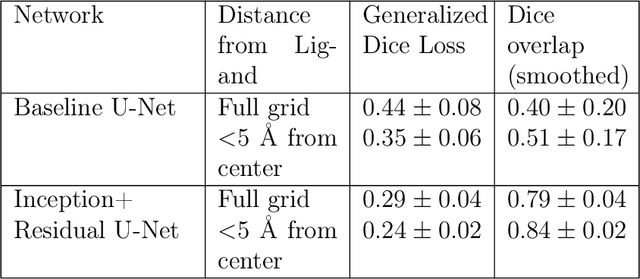
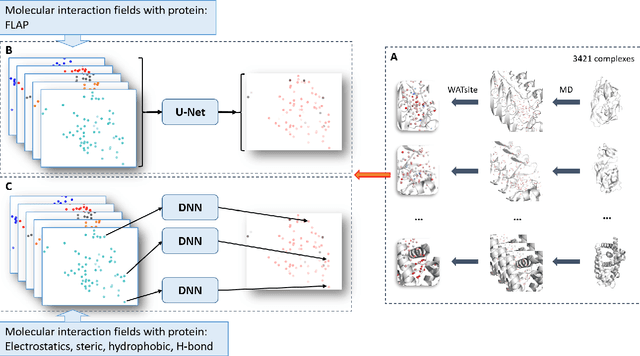
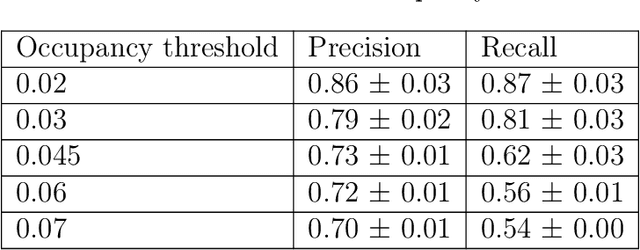
The calculation of thermodynamic properties of biochemical systems typically requires the use of resource-intensive molecular simulation methods. One example thereof is the thermodynamic profiling of hydration sites, i.e. high-probability locations for water molecules on the protein surface, which play an essential role in protein-ligand associations and must therefore be incorporated in the prediction of binding poses and affinities. To replace time-consuming simulations in hydration site predictions, we developed two different types of deep neural-network models aiming to predict hydration site data. In the first approach, meshed 3D images are generated representing the interactions between certain molecular probes placed on regular 3D grids, encompassing the binding pocket, with the static protein. These molecular interaction fields are mapped to the corresponding 3D image of hydration occupancy using a neural network based on an U-Net architecture. In a second approach, hydration occupancy and thermodynamics were predicted point-wise using a neural network based on fully-connected layers. In addition to direct protein interaction fields, the environment of each grid point was represented using moments of a spherical harmonics expansion of the interaction properties of nearby grid points. Application to structure-activity relationship analysis and protein-ligand pose scoring demonstrates the utility of the predicted hydration information.