 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultitask finetuning and acceleration of chemical pretrained models for small molecule drug property prediction
Oct 14, 2025Chemical pretrained models, sometimes referred to as foundation models, are receiving considerable interest for drug discovery applications. The general chemical knowledge extracted from self-supervised training has the potential to improve predictions for critical drug discovery endpoints, including on-target potency and ADMET properties. Multi-task learning has previously been successfully leveraged to improve predictive models. Here, we show that enabling multitasking in finetuning of chemical pretrained graph neural network models such as Kinetic GROVER Multi-Task (KERMT), an enhanced version of the GROVER model, and Knowledge-guided Pre-training of Graph Transformer (KGPT) significantly improves performance over non-pretrained graph neural network models. Surprisingly, we find that the performance improvement from finetuning KERMT in a multitask manner is most significant at larger data sizes. Additionally, we publish two multitask ADMET data splits to enable more accurate benchmarking of multitask deep learning methods for drug property prediction. Finally, we provide an accelerated implementation of the KERMT model on GitHub, unlocking large-scale pretraining, finetuning, and inference in industrial drug discovery workflows.
Step Change Improvement in ADMET Prediction with PotentialNet Deep Featurization
Mar 28, 2019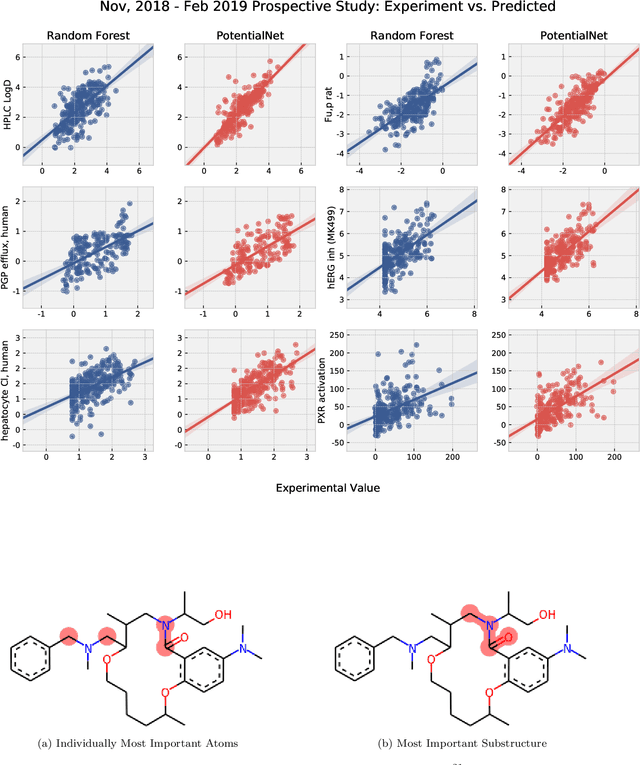
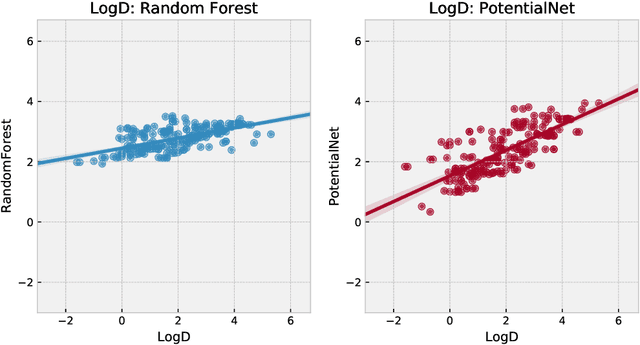
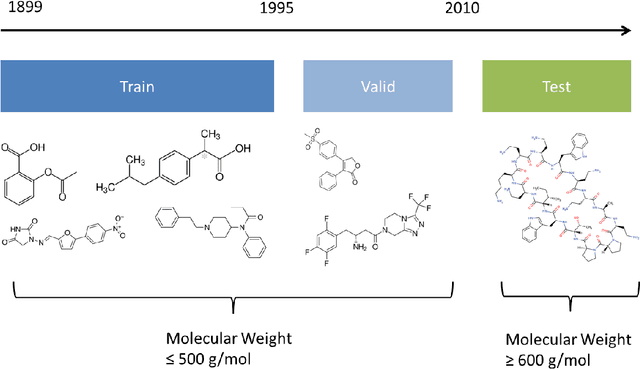
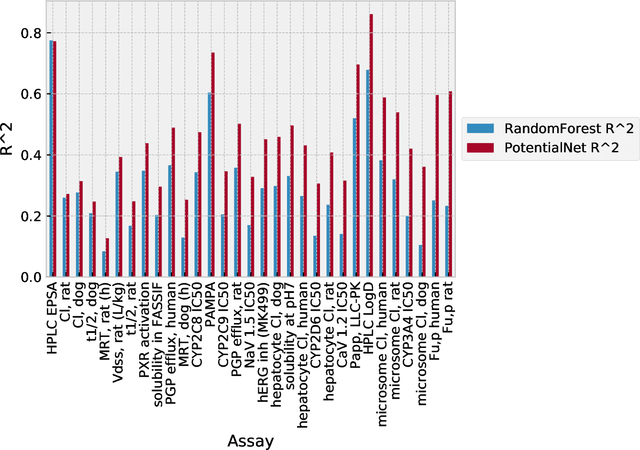
The Absorption, Distribution, Metabolism, Elimination, and Toxicity (ADMET) properties of drug candidates are estimated to account for up to 50% of all clinical trial failures. Predicting ADMET properties has therefore been of great interest to the cheminformatics and medicinal chemistry communities in recent decades. Traditional cheminformatics approaches, whether the learner is a random forest or a deep neural network, leverage fixed fingerprint feature representations of molecules. In contrast, in this paper, we learn the features most relevant to each chemical task at hand by representing each molecule explicitly as a graph, where each node is an atom and each edge is a bond. By applying graph convolutions to this explicit molecular representation, we achieve, to our knowledge, unprecedented accuracy in prediction of ADMET properties. By challenging our methodology with rigorous cross-validation procedures and prospective analyses, we show that deep featurization better enables molecular predictors to not only interpolate but also extrapolate to new regions of chemical space.