 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge"Information": models, code, and papers
Towards Unsupervised HPO for Outlier Detection
Aug 24, 2022
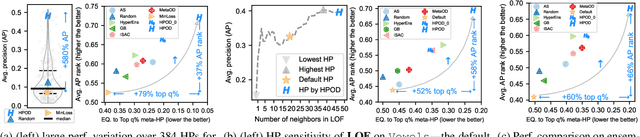

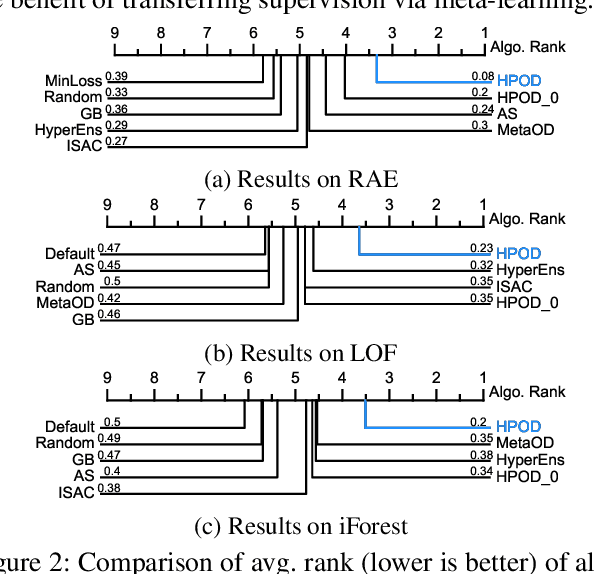
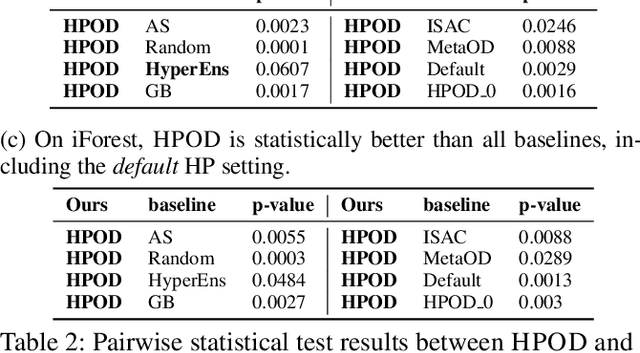
Given an unsupervised outlier detection (OD) algorithm, how can we optimize its hyperparameter(s) (HP) on a new dataset, without any labels? In this work, we address this challenging hyperparameter optimization for unsupervised OD problem, and propose the first systematic approach called HPOD that is based on meta-learning. HPOD capitalizes on the prior performance of a large collection of HPs on existing OD benchmark datasets, and transfers this information to enable HP evaluation on a new dataset without labels. Moreover, HPOD adapts (originally supervised) sequential model-based optimization to identify promising HPs efficiently. Extensive experiments show that HPOD works with both deep (e.g., Robust AutoEncoder) and shallow (e.g., Local Outlier Factor (LOF) and Isolation Forest (iForest)) OD algorithms on both discrete and continuous HP spaces, and outperforms a wide range of baselines with on average 58% and 66% performance improvement over the default HPs of LOF and iForest.
Characterizing Graph Datasets for Node Classification: Beyond Homophily-Heterophily Dichotomy
Sep 13, 2022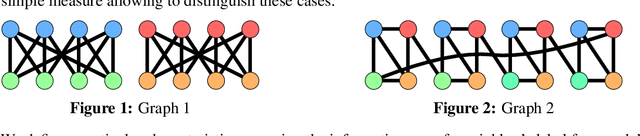
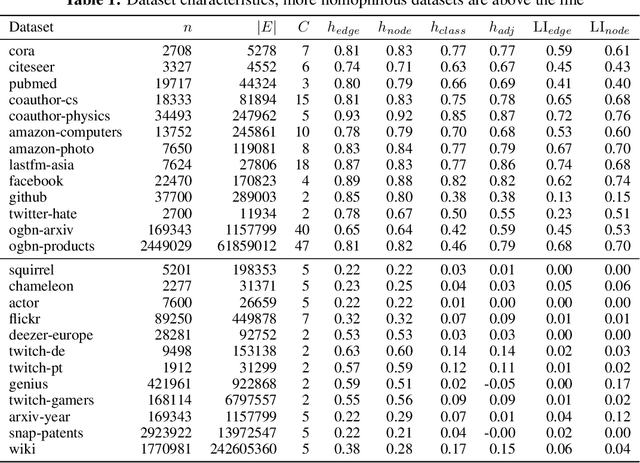
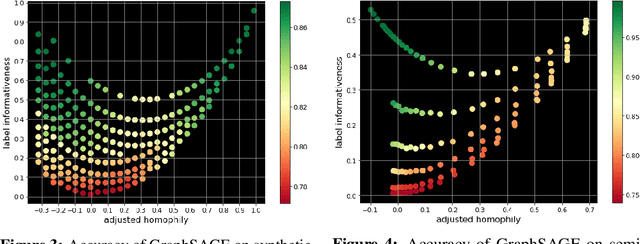
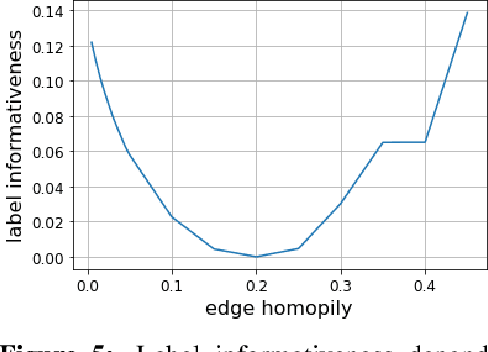
Homophily is a graph property describing the tendency of edges to connect similar nodes; the opposite is called heterophily. While homophily is natural for many real-world networks, there are also networks without this property. It is often believed that standard message-passing graph neural networks (GNNs) do not perform well on non-homophilous graphs, and thus such datasets need special attention. While a lot of effort has been put into developing graph representation learning methods for heterophilous graphs, there is no universally agreed upon measure of homophily. Several metrics for measuring homophily have been used in the literature, however, we show that all of them have critical drawbacks preventing comparison of homophily levels between different datasets. We formalize desirable properties for a proper homophily measure and show how existing literature on the properties of classification performance metrics can be linked to our problem. In doing so we find a measure that we call adjusted homophily that satisfies more desirable properties than existing homophily measures. Interestingly, this measure is related to two classification performance metrics - Cohen's Kappa and Matthews correlation coefficient. Then, we go beyond the homophily-heterophily dichotomy and propose a new property that we call label informativeness (LI) that characterizes how much information a neighbor's label provides about a node's label. We theoretically show that LI is comparable across datasets with different numbers of classes and class size balance. Through a series of experiments we show that LI is a better predictor of the performance of GNNs on a dataset than homophily. We show that LI explains why GNNs can sometimes perform well on heterophilous datasets - a phenomenon recently observed in the literature.
MHNF: Multi-hop Heterogeneous Neighborhood information Fusion graph representation learning
Jun 17, 2021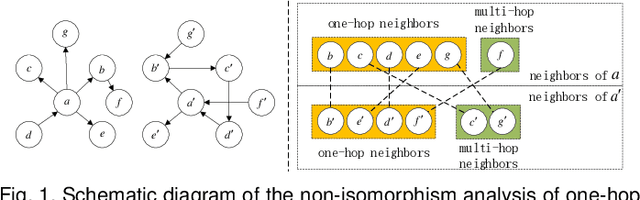

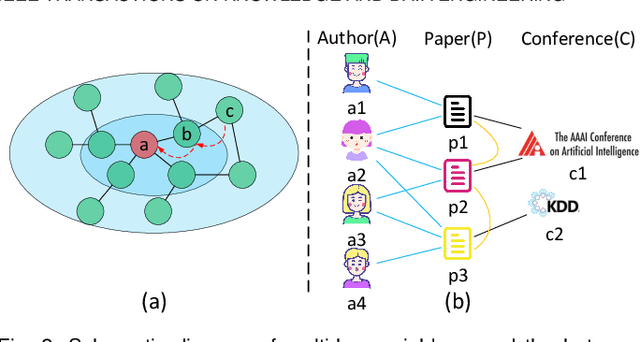
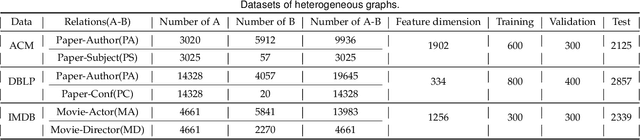
Attention mechanism enables the Graph Neural Networks(GNNs) to learn the attention weights between the target node and its one-hop neighbors, the performance is further improved. However, the most existing GNNs are oriented to homogeneous graphs and each layer can only aggregate the information of one-hop neighbors. Stacking multi-layer networks will introduce a lot of noise and easily lead to over smoothing. We propose a Multi-hop Heterogeneous Neighborhood information Fusion graph representation learning method (MHNF). Specifically, we first propose a hybrid metapath autonomous extraction model to efficiently extract multi-hop hybrid neighbors. Then, we propose a hop-level heterogeneous Information aggregation model, which selectively aggregates different-hop neighborhood information within the same hybrid metapath. Finally, a hierarchical semantic attention fusion model (HSAF) is proposed, which can efficiently integrate different-hop and different-path neighborhood information respectively. This paper can solve the problem of aggregating the multi-hop neighborhood information and can learn hybrid metapaths for target task, reducing the limitation of manually specifying metapaths. In addition, HSAF can extract the internal node information of the metapaths and better integrate the semantic information of different levels. Experimental results on real datasets show that MHNF is superior to state-of-the-art methods in node classification and clustering tasks (10.94% - 69.09% and 11.58% - 394.93% relative improvement on average, respectively).
Regional Differential Information Entropy for Super-Resolution Image Quality Assessment
Jul 08, 2021

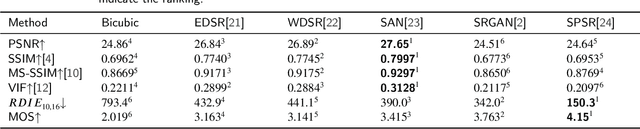

PSNR and SSIM are the most widely used metrics in super-resolution problems, because they are easy to use and can evaluate the similarities between generated images and reference images. However, single image super-resolution is an ill-posed problem, there are multiple corresponding high-resolution images for the same low-resolution image. The similarities can't totally reflect the restoration effect. The perceptual quality of generated images is also important, but PSNR and SSIM do not reflect perceptual quality well. To solve the problem, we proposed a method called regional differential information entropy to measure both of the similarities and perceptual quality. To overcome the problem that traditional image information entropy can't reflect the structure information, we proposed to measure every region's information entropy with sliding window. Considering that the human visual system is more sensitive to the brightness difference at low brightness, we take $\gamma$ quantization rather than linear quantization. To accelerate the method, we reorganized the calculation procedure of information entropy with a neural network. Through experiments on our IQA dataset and PIPAL, this paper proves that RDIE can better quantify perceptual quality of images especially GAN-based images.
Collaborative Algorithms for Online Personalized Mean Estimation
Aug 24, 2022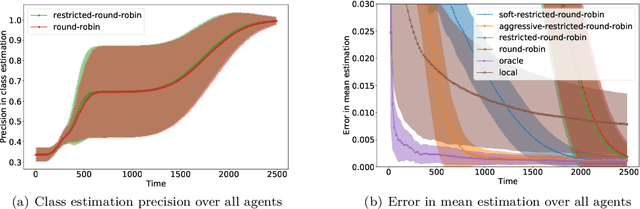

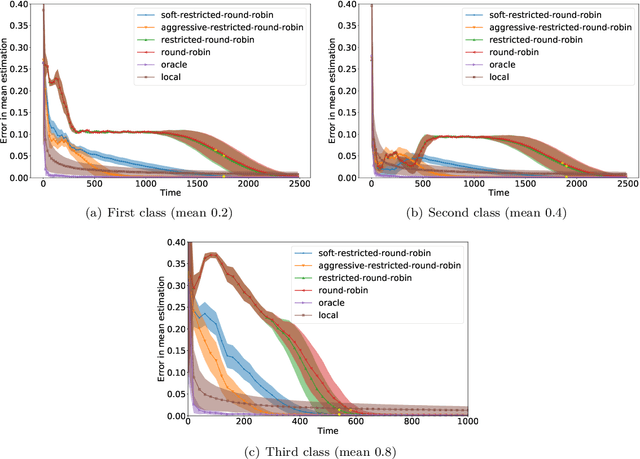
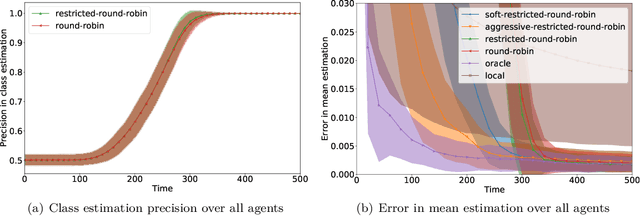
We consider an online estimation problem involving a set of agents. Each agent has access to a (personal) process that generates samples from a real-valued distribution and seeks to estimate its mean. We study the case where some of the distributions have the same mean, and the agents are allowed to actively query information from other agents. The goal is to design an algorithm that enables each agent to improve its mean estimate thanks to communication with other agents. The means as well as the number of distributions with same mean are unknown, which makes the task nontrivial. We introduce a novel collaborative strategy to solve this online personalized mean estimation problem. We analyze its time complexity and introduce variants that enjoy good performance in numerical experiments. We also extend our approach to the setting where clusters of agents with similar means seek to estimate the mean of their cluster.
Building a Chatbot on a Closed Domain using RASA
Aug 12, 2022
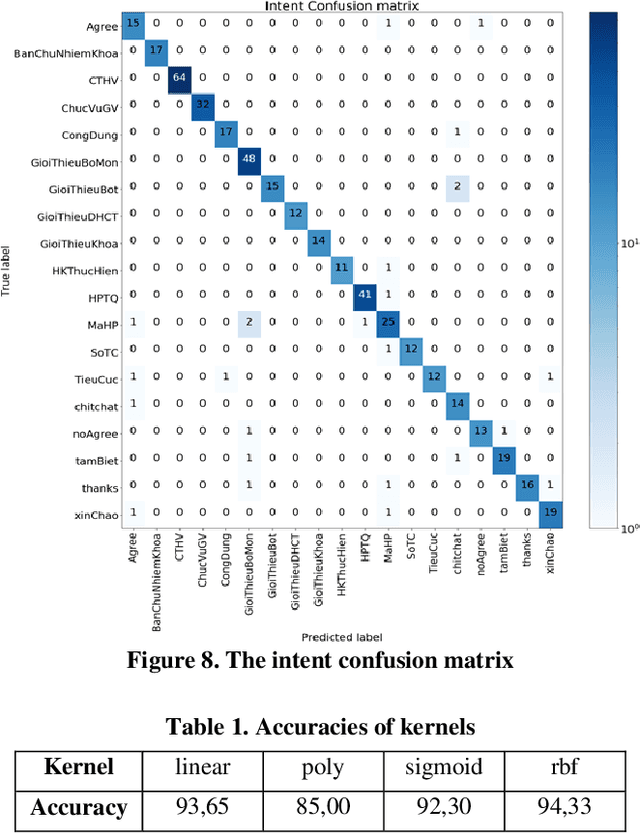

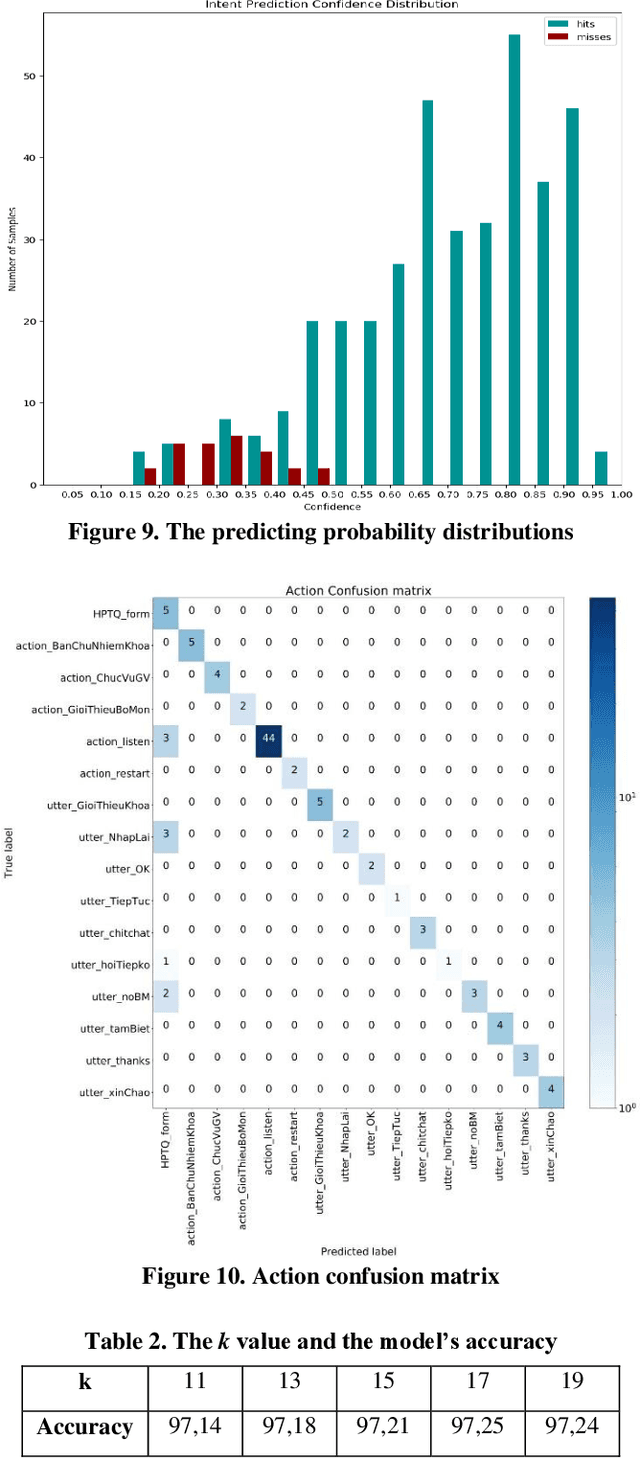
In this study, we build a chatbot system in a closed domain with the RASA framework, using several models such as SVM for classifying intents, CRF for extracting entities and LSTM for predicting action. To improve responses from the bot, the kNN algorithm is used to transform false entities extracted into true entities. The knowledge domain of our chatbot is about the College of Information and Communication Technology of Can Tho University, Vietnam. We manually construct a chatbot corpus with 19 intents, 441 sentence patterns of intents, 253 entities and 133 stories. Experiment results show that the bot responds well to relevant questions.
* 5 pages
Unified 2D and 3D Pre-Training of Molecular Representations
Jul 14, 2022
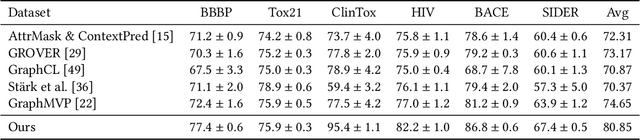
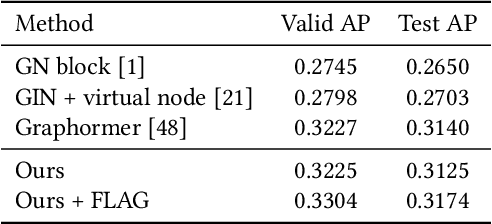
Molecular representation learning has attracted much attention recently. A molecule can be viewed as a 2D graph with nodes/atoms connected by edges/bonds, and can also be represented by a 3D conformation with 3-dimensional coordinates of all atoms. We note that most previous work handles 2D and 3D information separately, while jointly leveraging these two sources may foster a more informative representation. In this work, we explore this appealing idea and propose a new representation learning method based on a unified 2D and 3D pre-training. Atom coordinates and interatomic distances are encoded and then fused with atomic representations through graph neural networks. The model is pre-trained on three tasks: reconstruction of masked atoms and coordinates, 3D conformation generation conditioned on 2D graph, and 2D graph generation conditioned on 3D conformation. We evaluate our method on 11 downstream molecular property prediction tasks: 7 with 2D information only and 4 with both 2D and 3D information. Our method achieves state-of-the-art results on 10 tasks, and the average improvement on 2D-only tasks is 8.3%. Our method also achieves significant improvement on two 3D conformation generation tasks.
Computer vision based vehicle tracking as a complementary and scalable approach to RFID tagging
Sep 13, 2022
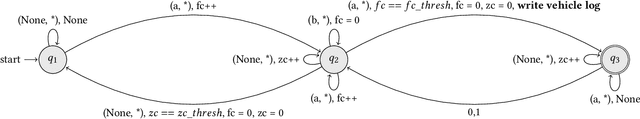


Logging of incoming/outgoing vehicles serves as a piece of critical information for root-cause analysis to combat security breach incidents in various sensitive organizations. RFID tagging hampers the scalability of vehicle tracking solutions on both logistics as well as technical fronts. For instance, requiring each incoming vehicle(departmental or private) to be RFID tagged is a severe constraint and coupling video analytics with RFID to detect abnormal vehicle movement is non-trivial. We leverage publicly available implementations of computer vision algorithms to develop an interpretable vehicle tracking algorithm using finite-state machine formalism. The state-machine consumes input from the cascaded object detection and optical character recognition(OCR) models for state transitions. We evaluated the proposed method on 75 video clips of 285 vehicles from our system deployment site. We observed that the detection rate is most affected by the speed and the type of vehicle. The highest detection rate is achieved when the vehicle movement is restricted to follow a movement restrictions(SOP) at the checkpoint similar to RFID tagging. We further analyzed 700 vehicle tracking predictions on live-data and identified that the majority of vehicle number prediction errors are due to illegible-text, image-blur, text occlusion and out-of-vocab letters in vehicle numbers. Towards system deployment and performance enhancement, we expect our ongoing system monitoring to provide evidences to establish a higher vehicle-throughput SOP at the security checkpoint as well as to drive the fine-tuning of the deployed computer-vision models and the state-machine to establish the proposed approach as a promising alternative to RFID-tagging.
TINC: Temporally Informed Non-Contrastive Learning for Disease Progression Modeling in Retinal OCT Volumes
Jun 30, 2022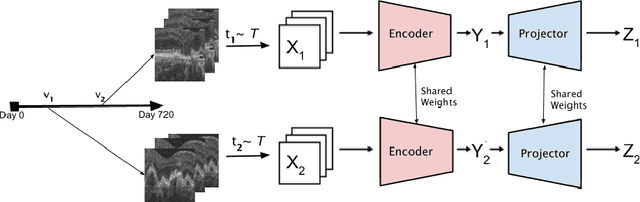

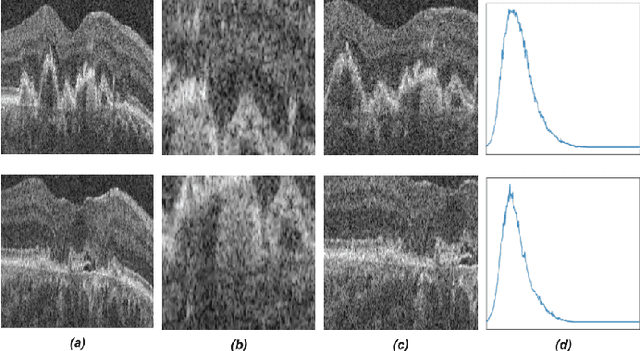

Recent contrastive learning methods achieved state-of-the-art in low label regimes. However, the training requires large batch sizes and heavy augmentations to create multiple views of an image. With non-contrastive methods, the negatives are implicitly incorporated in the loss, allowing different images and modalities as pairs. Although the meta-information (i.e., age, sex) in medical imaging is abundant, the annotations are noisy and prone to class imbalance. In this work, we exploited already existing temporal information (different visits from a patient) in a longitudinal optical coherence tomography (OCT) dataset using temporally informed non-contrastive loss (TINC) without increasing complexity and need for negative pairs. Moreover, our novel pair-forming scheme can avoid heavy augmentations and implicitly incorporates the temporal information in the pairs. Finally, these representations learned from the pretraining are more successful in predicting disease progression where the temporal information is crucial for the downstream task. More specifically, our model outperforms existing models in predicting the risk of conversion within a time frame from intermediate age-related macular degeneration (AMD) to the late wet-AMD stage.
HelixFold-Single: MSA-free Protein Structure Prediction by Using Protein Language Model as an Alternative
Aug 09, 2022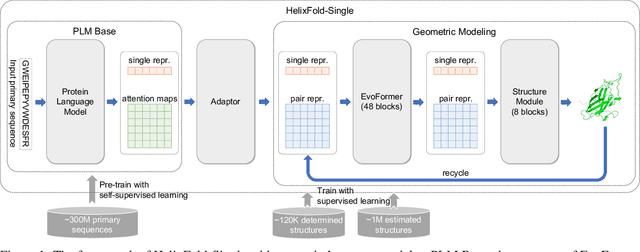

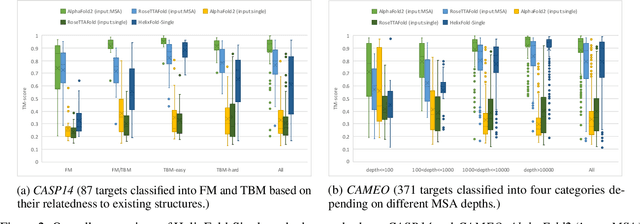
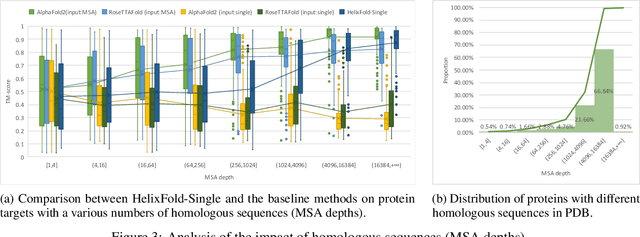
AI-based protein structure prediction pipelines, such as AlphaFold2, have achieved near-experimental accuracy. These advanced pipelines mainly rely on Multiple Sequence Alignments (MSAs) as inputs to learn the co-evolution information from the homologous sequences. Nonetheless, searching MSAs from protein databases is time-consuming, usually taking dozens of minutes. Consequently, we attempt to explore the limits of fast protein structure prediction by using only primary sequences of proteins. HelixFold-Single is proposed to combine a large-scale protein language model with the superior geometric learning capability of AlphaFold2. Our proposed method, HelixFold-Single, first pre-trains a large-scale protein language model (PLM) with thousands of millions of primary sequences utilizing the self-supervised learning paradigm, which will be used as an alternative to MSAs for learning the co-evolution information. Then, by combining the pre-trained PLM and the essential components of AlphaFold2, we obtain an end-to-end differentiable model to predict the 3D coordinates of atoms from only the primary sequence. HelixFold-Single is validated in datasets CASP14 and CAMEO, achieving competitive accuracy with the MSA-based methods on the targets with large homologous families. Furthermore, HelixFold-Single consumes much less time than the mainstream pipelines for protein structure prediction, demonstrating its potential in tasks requiring many predictions. The code of HelixFold-Single is available at https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold-single, and we also provide stable web services on https://paddlehelix.baidu.com/app/drug/protein-single/forecast.