 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDoes GNN Pretraining Help Molecular Representation?
Paper and Code
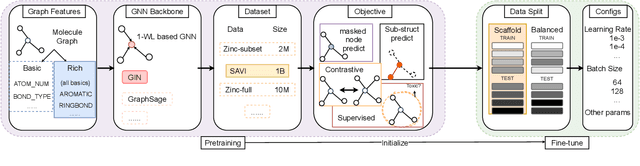
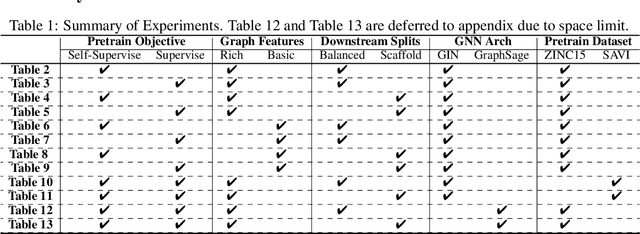
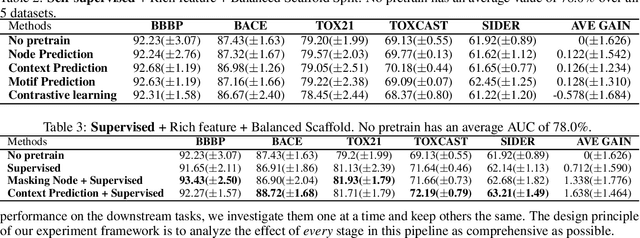
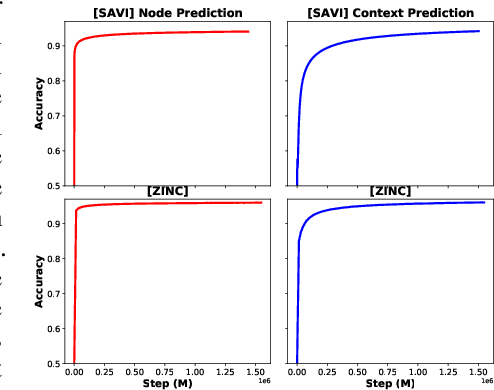
Extracting informative representations of molecules using Graph neural networks (GNNs) is crucial in AI-driven drug discovery. Recently, the graph research community has been trying to replicate the success of self-supervised pretraining in natural language processing, with several successes claimed. However, we find the benefit brought by self-supervised pretraining on molecular data can be negligible in many cases. We conduct thorough ablation studies on the key components of GNN pretraining, including pretraining objectives, data splitting methods, input features, pretraining dataset scales, and GNN architectures, in deciding the accuracy of the downstream tasks. Our first important finding is, self-supervised graph pretraining do not have statistically significant advantages over non-pretraining methods in many settings. Second, although improvement can be observed with additional supervised pretraining, the improvement may diminish with richer features or more balanced data splits. Third, experimental hyper-parameters have a larger impact on accuracy of downstream tasks than the choice of pretraining tasks. We hypothesize the complexity of pretraining on molecules is insufficient, leading to less transferable knowledge for downstream tasks.