 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMeaningful machine learning models and machine-learned pharmacophores from fragment screening campaigns
Paper and Code
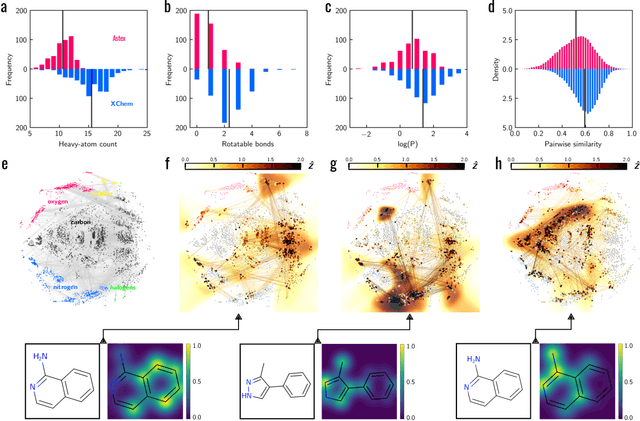
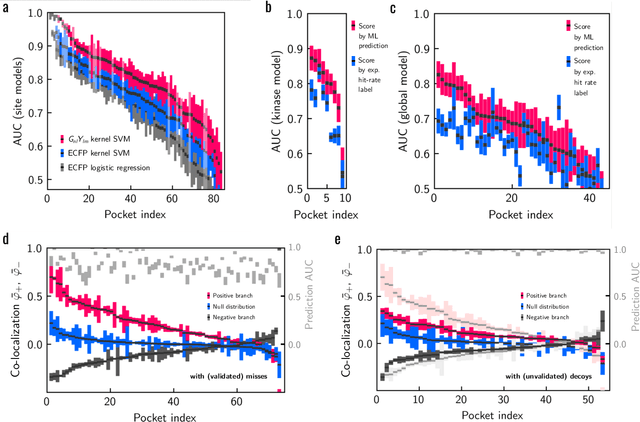
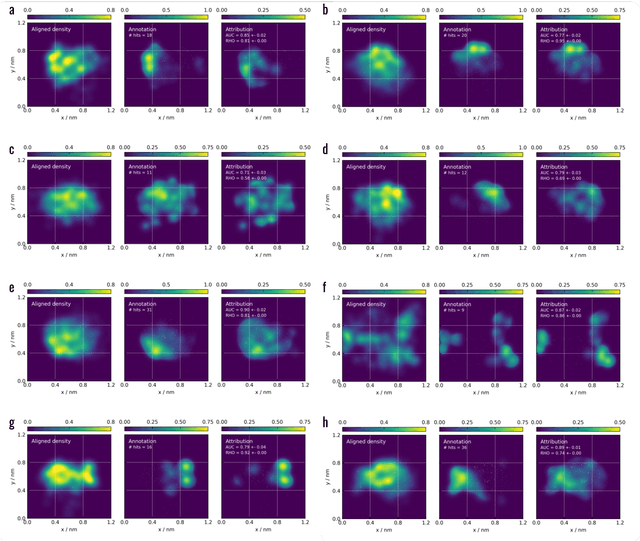
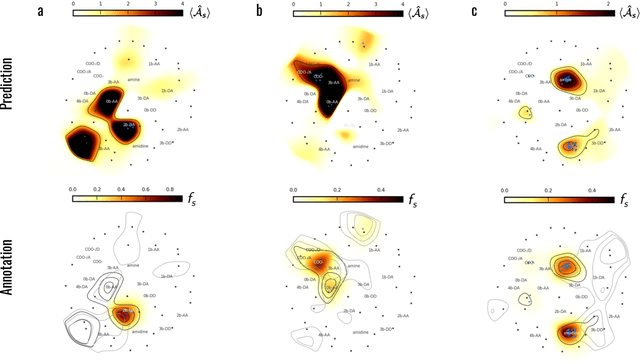
Machine learning (ML) is widely used in drug discovery to train models that predict protein-ligand binding. These models are of great value to medicinal chemists, in particular if they provide case-specific insight into the physical interactions that drive the binding process. In this study we derive ML models from over 50 fragment-screening campaigns to introduce two important elements that we believe are absent in most -- if not all -- ML studies of this type reported to date: First, alongside the observed hits we use to train our models, we incorporate true misses and show that these experimentally validated negative data are of significant importance to the quality of the derived models. Second, we provide a physically interpretable and verifiable representation of what the ML model considers important for successful binding. This representation is derived from a straightforward attribution procedure that explains the prediction in terms of the (inter-)action of chemical environments. Critically, we validate the attribution outcome on a large scale against prior annotations made independently by expert molecular modellers. We find good agreement between the key molecular substructures proposed by the ML model and those assigned manually, even when the model's performance in discriminating hits from misses is far from perfect. By projecting the attribution onto predefined interaction prototypes (pharmacophores), we show that ML allows us to formulate simple rules for what drives fragment binding against a target automatically from screening data.