 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular representation learning with language models and domain-relevant auxiliary tasks
Paper and Code
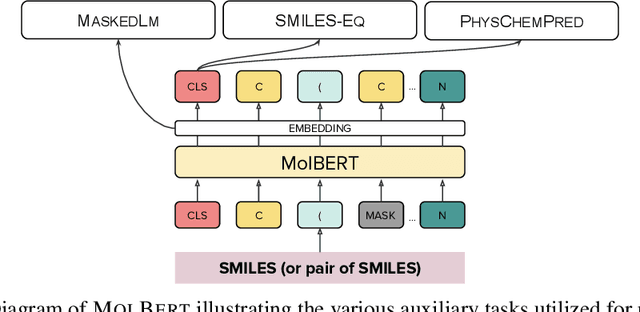

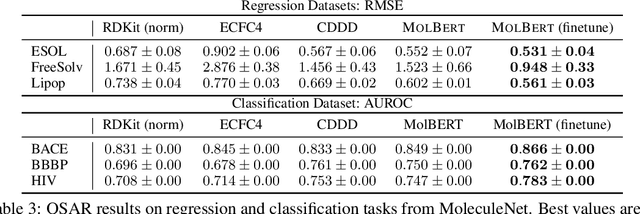
We apply a Transformer architecture, specifically BERT, to learn flexible and high quality molecular representations for drug discovery problems. We study the impact of using different combinations of self-supervised tasks for pre-training, and present our results for the established Virtual Screening and QSAR benchmarks. We show that: i) The selection of appropriate self-supervised task(s) for pre-training has a significant impact on performance in subsequent downstream tasks such as Virtual Screening. ii) Using auxiliary tasks with more domain relevance for Chemistry, such as learning to predict calculated molecular properties, increases the fidelity of our learnt representations. iii) Finally, we show that molecular representations learnt by our model `MolBert' improve upon the current state of the art on the benchmark datasets.