 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning mutational graphs of individual tumor evolution from multi-sample sequencing data
Paper and Code
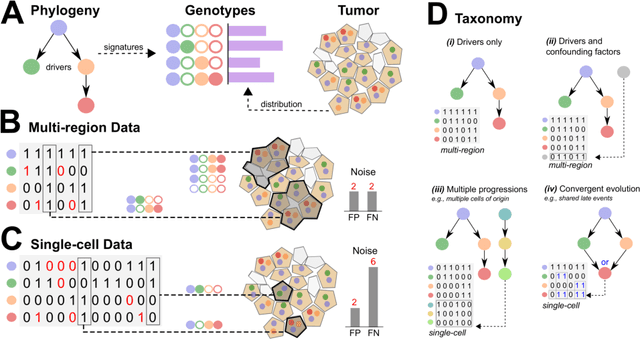
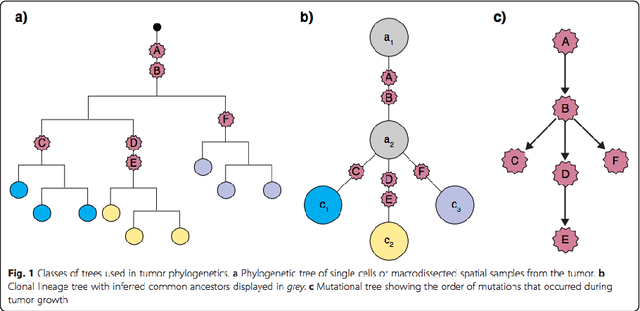
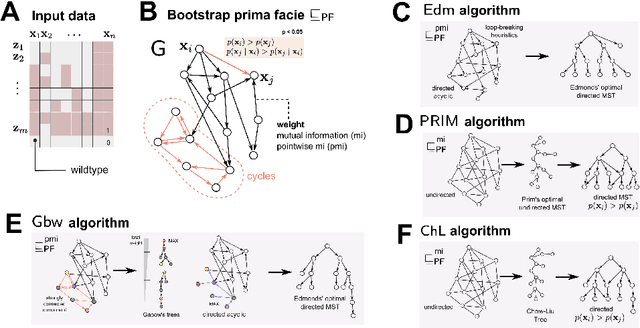
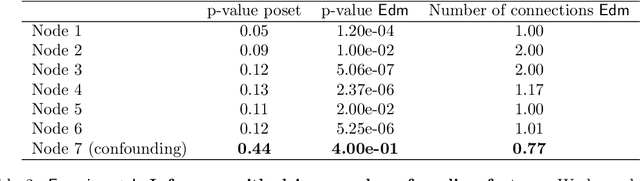
Phylogenetic techniques quantify intra-tumor heterogeneity by deconvolving either clonal or mutational trees from multi-sample sequencing data of individual tumors. Most of these methods rely on the well-known infinite sites assumption, and are limited to process either multi-region or single-cell sequencing data. Here, we improve over those methods with TRaIT, a unified statistical framework for the inference of the accumula- tion order of multiple types of genomic alterations driving tumor development. TRaIT supports both multi-region and single-cell sequencing data, and output mutational graphs accounting for violations of the infinite sites assumption due to convergent evolution, and other complex phenomena that cannot be detected with phylogenetic tools. Our method displays better accuracy, performance and robustness to noise and small sample size than state-of-the-art phylogenetic methods. We show with single-cell data from breast cancer and multi-region data from colorectal cancer that TRaIT can quantify the extent of intra-tumor heterogeneity and generate new testable experimental hypotheses.