 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeYuankai Huo
An Accelerated Pipeline for Multi-label Renal Pathology Image Segmentation at the Whole Slide Image Level
May 23, 2023
Deep-learning techniques have been used widely to alleviate the labour-intensive and time-consuming manual annotation required for pixel-level tissue characterization. Our previous study introduced an efficient single dynamic network - Omni-Seg - that achieved multi-class multi-scale pathological segmentation with less computational complexity. However, the patch-wise segmentation paradigm still applies to Omni-Seg, and the pipeline is time-consuming when providing segmentation for Whole Slide Images (WSIs). In this paper, we propose an enhanced version of the Omni-Seg pipeline in order to reduce the repetitive computing processes and utilize a GPU to accelerate the model's prediction for both better model performance and faster speed. Our proposed method's innovative contribution is two-fold: (1) a Docker is released for an end-to-end slide-wise multi-tissue segmentation for WSIs; and (2) the pipeline is deployed on a GPU to accelerate the prediction, achieving better segmentation quality in less time. The proposed accelerated implementation reduced the average processing time (at the testing stage) on a standard needle biopsy WSI from 2.3 hours to 22 minutes, using 35 WSIs from the Kidney Tissue Atlas (KPMP) Datasets. The source code and the Docker have been made publicly available at https://github.com/ddrrnn123/Omni-Seg.
An End-to-end Pipeline for 3D Slide-wise Multi-stain Renal Pathology Registration
May 19, 2023Tissue examination and quantification in a 3D context on serial section whole slide images (WSIs) were laborintensive and time-consuming tasks. Our previous study proposed a novel registration-based method (Map3D) to automatically align WSIs to the same physical space, reducing the human efforts of screening serial sections from WSIs. However, the registration performance of our Map3D method was only evaluated on single-stain WSIs with large-scale kidney tissue samples. In this paper, we provide a Docker for an end-to-end 3D slide-wise registration pipeline on needle biopsy serial sections in a multi-stain paradigm. The contribution of this study is three-fold: (1) We release a containerized Docker for an end-to-end multi-stain WSI registration. (2) We prove that the Map3D pipeline is capable of sectional registration from multi-stain WSI. (3) We verify that the Map3D pipeline can also be applied to needle biopsy tissue samples. The source code and the Docker have been made publicly available at https://github.com/hrlblab/Map3D.
* 6 pages, 4 figures
Exploring shared memory architectures for end-to-end gigapixel deep learning
Apr 24, 2023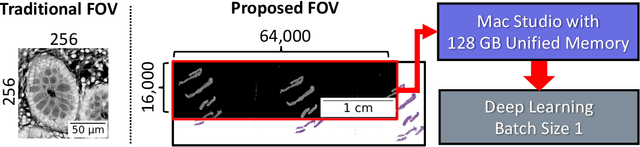

Deep learning has made great strides in medical imaging, enabled by hardware advances in GPUs. One major constraint for the development of new models has been the saturation of GPU memory resources during training. This is especially true in computational pathology, where images regularly contain more than 1 billion pixels. These pathological images are traditionally divided into small patches to enable deep learning due to hardware limitations. In this work, we explore whether the shared GPU/CPU memory architecture on the M1 Ultra systems-on-a-chip (SoCs) recently released by Apple, Inc. may provide a solution. These affordable systems (less than \$5000) provide access to 128 GB of unified memory (Mac Studio with M1 Ultra SoC). As a proof of concept for gigapixel deep learning, we identified tissue from background on gigapixel areas from whole slide images (WSIs). The model was a modified U-Net (4492 parameters) leveraging large kernels and high stride. The M1 Ultra SoC was able to train the model directly on gigapixel images (16000$\times$64000 pixels, 1.024 billion pixels) with a batch size of 1 using over 100 GB of unified memory for the process at an average speed of 1 minute and 21 seconds per batch with Tensorflow 2/Keras. As expected, the model converged with a high Dice score of 0.989 $\pm$ 0.005. Training up until this point took 111 hours and 24 minutes over 4940 steps. Other high RAM GPUs like the NVIDIA A100 (largest commercially accessible at 80 GB, $\sim$\$15000) are not yet widely available (in preview for select regions on Amazon Web Services at \$40.96/hour as a group of 8). This study is a promising step towards WSI-wise end-to-end deep learning with prevalent network architectures.
Segment Anything Model (SAM) for Digital Pathology: Assess Zero-shot Segmentation on Whole Slide Imaging
Apr 09, 2023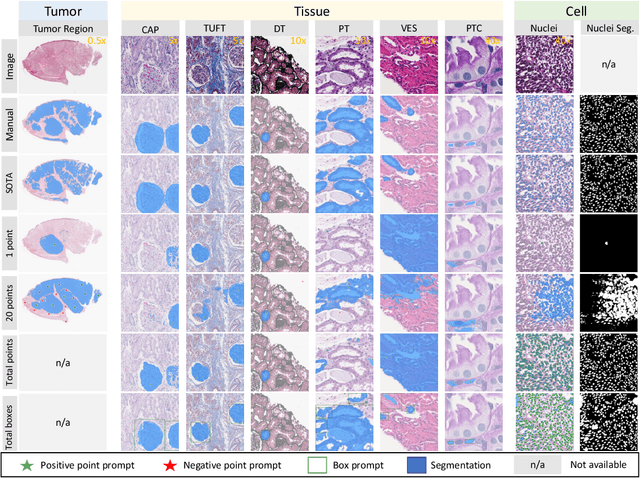
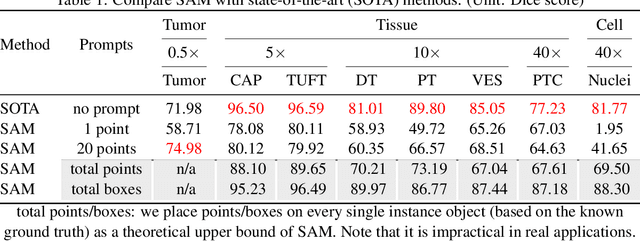
The segment anything model (SAM) was released as a foundation model for image segmentation. The promptable segmentation model was trained by over 1 billion masks on 11M licensed and privacy-respecting images. The model supports zero-shot image segmentation with various segmentation prompts (e.g., points, boxes, masks). It makes the SAM attractive for medical image analysis, especially for digital pathology where the training data are rare. In this study, we evaluate the zero-shot segmentation performance of SAM model on representative segmentation tasks on whole slide imaging (WSI), including (1) tumor segmentation, (2) non-tumor tissue segmentation, (3) cell nuclei segmentation. Core Results: The results suggest that the zero-shot SAM model achieves remarkable segmentation performance for large connected objects. However, it does not consistently achieve satisfying performance for dense instance object segmentation, even with 20 prompts (clicks/boxes) on each image. We also summarized the identified limitations for digital pathology: (1) image resolution, (2) multiple scales, (3) prompt selection, and (4) model fine-tuning. In the future, the few-shot fine-tuning with images from downstream pathological segmentation tasks might help the model to achieve better performance in dense object segmentation.
Zero-shot CT Field-of-view Completion with Unconditional Generative Diffusion Prior
Apr 07, 2023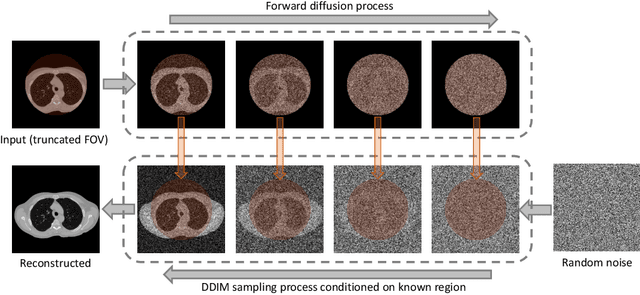
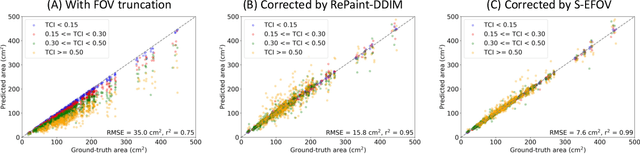
Anatomically consistent field-of-view (FOV) completion to recover truncated body sections has important applications in quantitative analyses of computed tomography (CT) with limited FOV. Existing solution based on conditional generative models relies on the fidelity of synthetic truncation patterns at training phase, which poses limitations for the generalizability of the method to potential unknown types of truncation. In this study, we evaluate a zero-shot method based on a pretrained unconditional generative diffusion prior, where truncation pattern with arbitrary forms can be specified at inference phase. In evaluation on simulated chest CT slices with synthetic FOV truncation, the method is capable of recovering anatomically consistent body sections and subcutaneous adipose tissue measurement error caused by FOV truncation. However, the correction accuracy is inferior to the conditionally trained counterpart.
Cross-scale Multi-instance Learning for Pathological Image Diagnosis
Apr 01, 2023
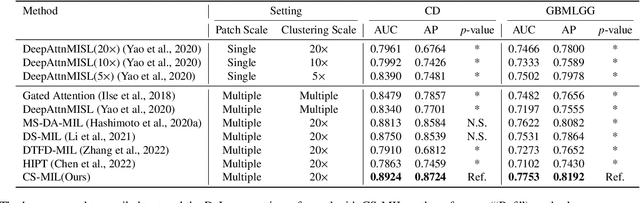
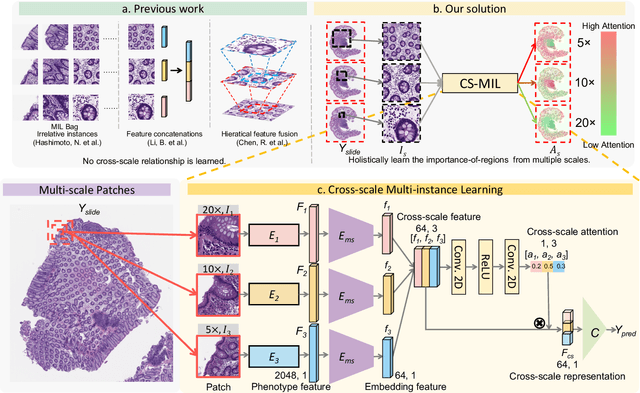

Analyzing high resolution whole slide images (WSIs) with regard to information across multiple scales poses a significant challenge in digital pathology. Multi-instance learning (MIL) is a common solution for working with high resolution images by classifying bags of objects (i.e. sets of smaller image patches). However, such processing is typically performed at a single scale (e.g., 20x magnification) of WSIs, disregarding the vital inter-scale information that is key to diagnoses by human pathologists. In this study, we propose a novel cross-scale MIL algorithm to explicitly aggregate inter-scale relationships into a single MIL network for pathological image diagnosis. The contribution of this paper is three-fold: (1) A novel cross-scale MIL (CS-MIL) algorithm that integrates the multi-scale information and the inter-scale relationships is proposed; (2) A toy dataset with scale-specific morphological features is created and released to examine and visualize differential cross-scale attention; (3) Superior performance on both in-house and public datasets is demonstrated by our simple cross-scale MIL strategy. The official implementation is publicly available at https://github.com/hrlblab/CS-MIL.
A Unified Single-stage Learning Model for Estimating Fiber Orientation Distribution Functions on Heterogeneous Multi-shell Diffusion-weighted MRI
Mar 29, 2023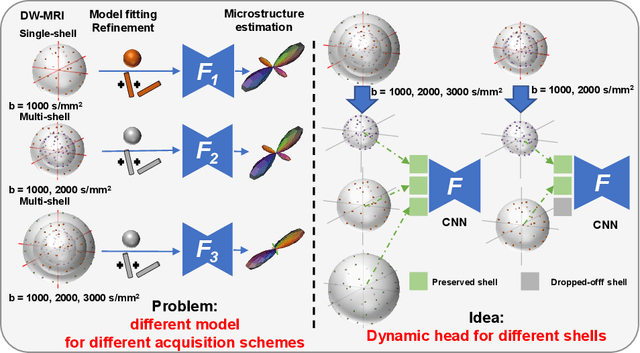
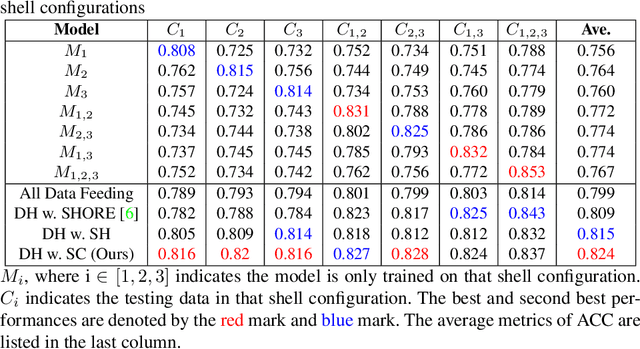

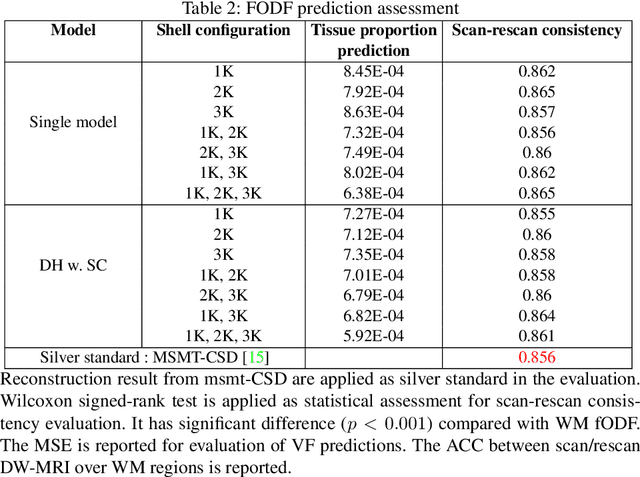
Diffusion-weighted (DW) MRI measures the direction and scale of the local diffusion process in every voxel through its spectrum in q-space, typically acquired in one or more shells. Recent developments in micro-structure imaging and multi-tissue decomposition have sparked renewed attention to the radial b-value dependence of the signal. Applications in tissue classification and micro-architecture estimation, therefore, require a signal representation that extends over the radial as well as angular domain. Multiple approaches have been proposed that can model the non-linear relationship between the DW-MRI signal and biological microstructure. In the past few years, many deep learning-based methods have been developed towards faster inference speed and higher inter-scan consistency compared with traditional model-based methods (e.g., multi-shell multi-tissue constrained spherical deconvolution). However, a multi-stage learning strategy is typically required since the learning process relied on various middle representations, such as simple harmonic oscillator reconstruction (SHORE) representation. In this work, we present a unified dynamic network with a single-stage spherical convolutional neural network, which allows efficient fiber orientation distribution function (fODF) estimation through heterogeneous multi-shell diffusion MRI sequences. We study the Human Connectome Project (HCP) young adults with test-retest scans. From the experimental results, the proposed single-stage method outperforms prior multi-stage approaches in repeated fODF estimation with shell dropoff and single-shell DW-MRI sequences.
Scaling Up 3D Kernels with Bayesian Frequency Re-parameterization for Medical Image Segmentation
Mar 10, 2023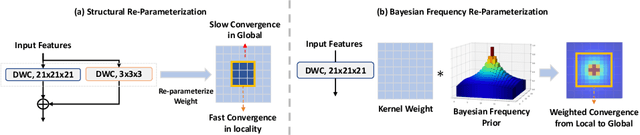
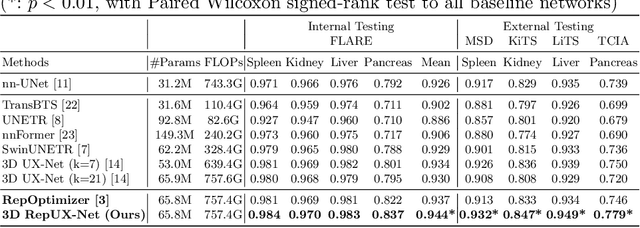
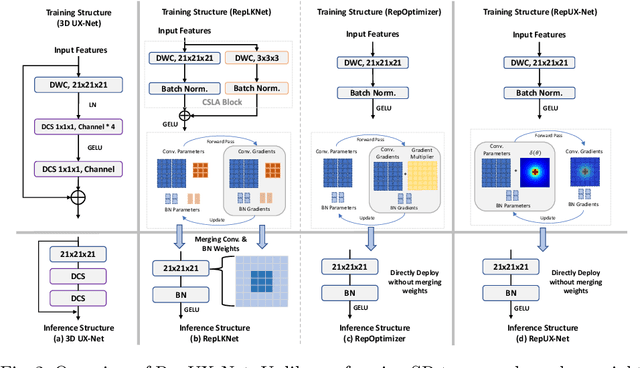

With the inspiration of vision transformers, the concept of depth-wise convolution revisits to provide a large Effective Receptive Field (ERF) using Large Kernel (LK) sizes for medical image segmentation. However, the segmentation performance might be saturated and even degraded as the kernel sizes scaled up (e.g., $21\times 21\times 21$) in a Convolutional Neural Network (CNN). We hypothesize that convolution with LK sizes is limited to maintain an optimal convergence for locality learning. While Structural Re-parameterization (SR) enhances the local convergence with small kernels in parallel, optimal small kernel branches may hinder the computational efficiency for training. In this work, we propose RepUX-Net, a pure CNN architecture with a simple large kernel block design, which competes favorably with current network state-of-the-art (SOTA) (e.g., 3D UX-Net, SwinUNETR) using 6 challenging public datasets. We derive an equivalency between kernel re-parameterization and the branch-wise variation in kernel convergence. Inspired by the spatial frequency in the human visual system, we extend to vary the kernel convergence into element-wise setting and model the spatial frequency as a Bayesian prior to re-parameterize convolutional weights during training. Specifically, a reciprocal function is leveraged to estimate a frequency-weighted value, which rescales the corresponding kernel element for stochastic gradient descent. From the experimental results, RepUX-Net consistently outperforms 3D SOTA benchmarks with internal validation (FLARE: 0.929 to 0.944), external validation (MSD: 0.901 to 0.932, KiTS: 0.815 to 0.847, LiTS: 0.933 to 0.949, TCIA: 0.736 to 0.779) and transfer learning (AMOS: 0.880 to 0.911) scenarios in Dice Score.
Single Slice Thigh CT Muscle Group Segmentation with Domain Adaptation and Self-Training
Nov 30, 2022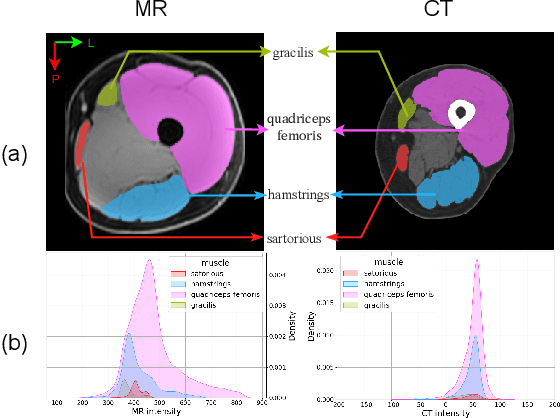
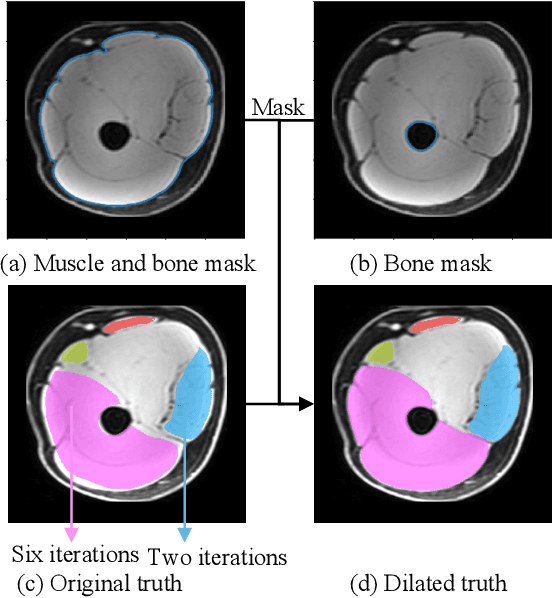
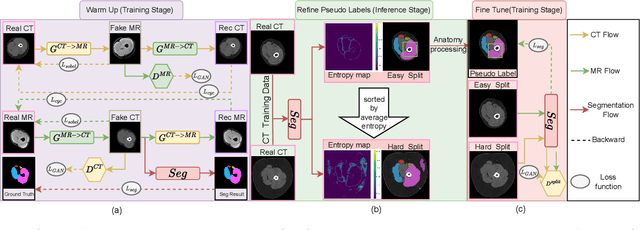
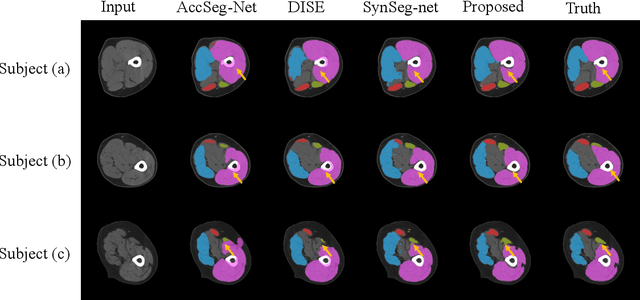
Objective: Thigh muscle group segmentation is important for assessment of muscle anatomy, metabolic disease and aging. Many efforts have been put into quantifying muscle tissues with magnetic resonance (MR) imaging including manual annotation of individual muscles. However, leveraging publicly available annotations in MR images to achieve muscle group segmentation on single slice computed tomography (CT) thigh images is challenging. Method: We propose an unsupervised domain adaptation pipeline with self-training to transfer labels from 3D MR to single CT slice. First, we transform the image appearance from MR to CT with CycleGAN and feed the synthesized CT images to a segmenter simultaneously. Single CT slices are divided into hard and easy cohorts based on the entropy of pseudo labels inferenced by the segmenter. After refining easy cohort pseudo labels based on anatomical assumption, self-training with easy and hard splits is applied to fine tune the segmenter. Results: On 152 withheld single CT thigh images, the proposed pipeline achieved a mean Dice of 0.888(0.041) across all muscle groups including sartorius, hamstrings, quadriceps femoris and gracilis. muscles Conclusion: To our best knowledge, this is the first pipeline to achieve thigh imaging domain adaptation from MR to CT. The proposed pipeline is effective and robust in extracting muscle groups on 2D single slice CT thigh images.The container is available for public use at https://github.com/MASILab/DA_CT_muscle_seg
CircleSnake: Instance Segmentation with Circle Representation
Nov 02, 2022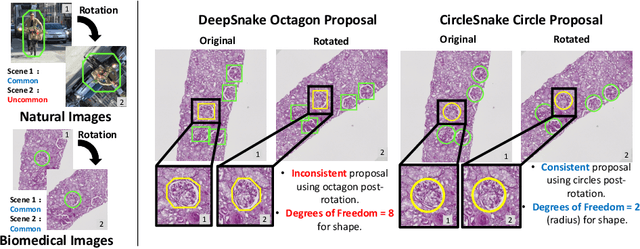
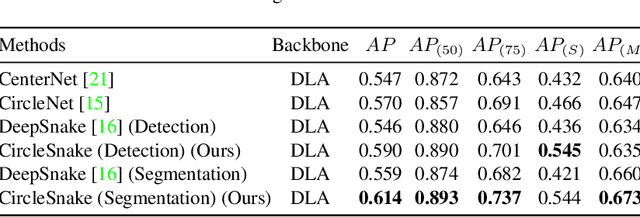
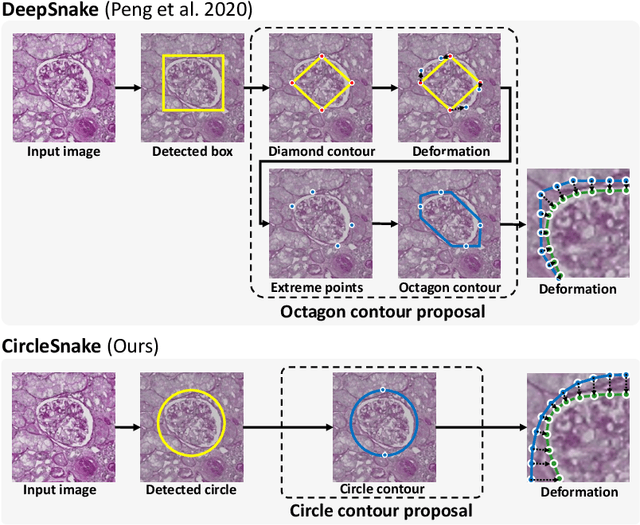
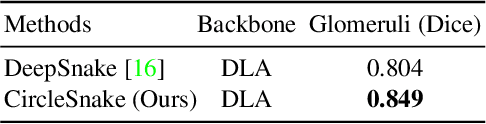
Circle representation has recently been introduced as a medical imaging optimized representation for more effective instance object detection on ball-shaped medical objects. With its superior performance on instance detection, it is appealing to extend the circle representation to instance medical object segmentation. In this work, we propose CircleSnake, a simple end-to-end circle contour deformation-based segmentation method for ball-shaped medical objects. Compared to the prevalent DeepSnake method, our contribution is three-fold: (1) We replace the complicated bounding box to octagon contour transformation with a computation-free and consistent bounding circle to circle contour adaption for segmenting ball-shaped medical objects; (2) Circle representation has fewer degrees of freedom (DoF=2) as compared with the octagon representation (DoF=8), thus yielding a more robust segmentation performance and better rotation consistency; (3) To the best of our knowledge, the proposed CircleSnake method is the first end-to-end circle representation deep segmentation pipeline method with consistent circle detection, circle contour proposal, and circular convolution. The key innovation is to integrate the circular graph convolution with circle detection into an end-to-end instance segmentation framework, enabled by the proposed simple and consistent circle contour representation. Glomeruli are used to evaluate the performance of the benchmarks. From the results, CircleSnake increases the average precision of glomerular detection from 0.559 to 0.614. The Dice score increased from 0.804 to 0.849. The code has been released: https://github.com/hrlblab/CircleSnake