 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStan Z. Li
LongVQ: Long Sequence Modeling with Vector Quantization on Structured Memory
Apr 18, 2024
Transformer models have been successful in various sequence processing tasks, but the self-attention mechanism's computational cost limits its practicality for long sequences. Although there are existing attention variants that improve computational efficiency, they have a limited ability to abstract global information effectively based on their hand-crafted mixing strategies. On the other hand, state-space models (SSMs) are tailored for long sequences but cannot capture complicated local information. Therefore, the combination of them as a unified token mixer is a trend in recent long-sequence models. However, the linearized attention degrades performance significantly even when equipped with SSMs. To address the issue, we propose a new method called LongVQ. LongVQ uses the vector quantization (VQ) technique to compress the global abstraction as a length-fixed codebook, enabling the linear-time computation of the attention matrix. This technique effectively maintains dynamic global and local patterns, which helps to complement the lack of long-range dependency issues. Our experiments on the Long Range Arena benchmark, autoregressive language modeling, and image and speech classification demonstrate the effectiveness of LongVQ. Our model achieves significant improvements over other sequence models, including variants of Transformers, Convolutions, and recent State Space Models.
AdaNovo: Adaptive \emph{De Novo} Peptide Sequencing with Conditional Mutual Information
Mar 15, 2024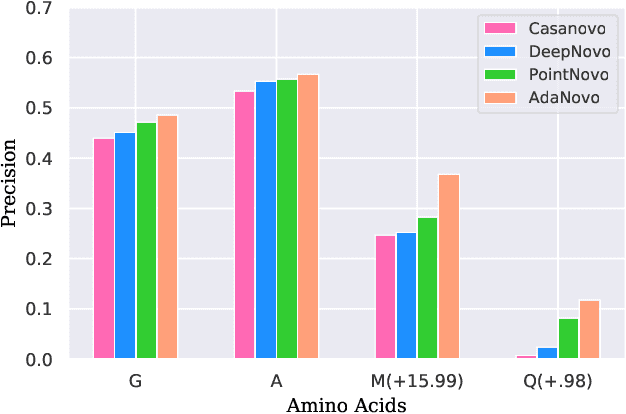
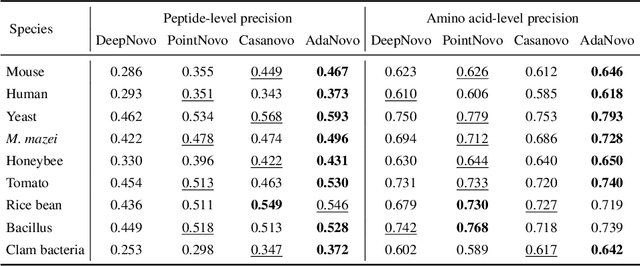
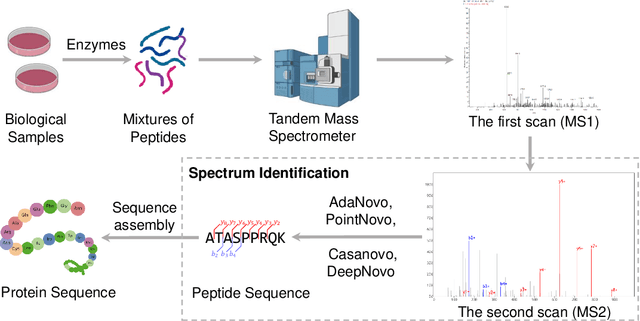
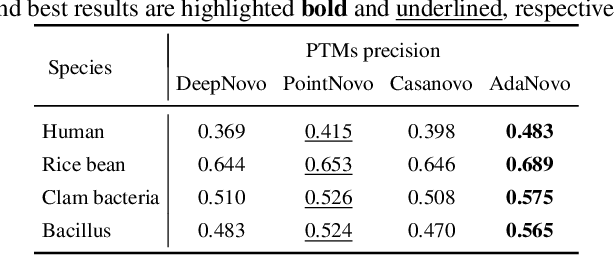
Tandem mass spectrometry has played a pivotal role in advancing proteomics, enabling the analysis of protein composition in biological samples. Despite the development of various deep learning methods for identifying amino acid sequences (peptides) responsible for observed spectra, challenges persist in \emph{de novo} peptide sequencing. Firstly, prior methods struggle to identify amino acids with post-translational modifications (PTMs) due to their lower frequency in training data compared to canonical amino acids, further resulting in decreased peptide-level identification precision. Secondly, diverse types of noise and missing peaks in mass spectra reduce the reliability of training data (peptide-spectrum matches, PSMs). To address these challenges, we propose AdaNovo, a novel framework that calculates conditional mutual information (CMI) between the spectrum and each amino acid/peptide, using CMI for adaptive model training. Extensive experiments demonstrate AdaNovo's state-of-the-art performance on a 9-species benchmark, where the peptides in the training set are almost completely disjoint from the peptides of the test sets. Moreover, AdaNovo excels in identifying amino acids with PTMs and exhibits robustness against data noise. The supplementary materials contain the official code.
A Teacher-Free Graph Knowledge Distillation Framework with Dual Self-Distillation
Mar 06, 2024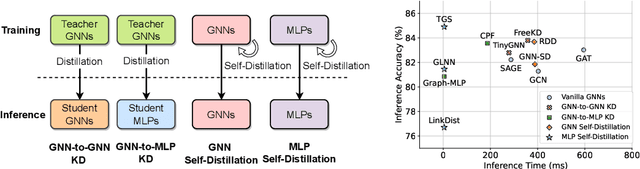
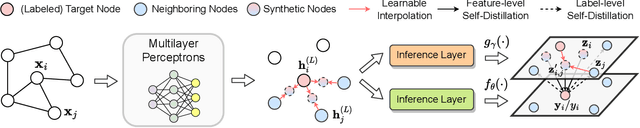
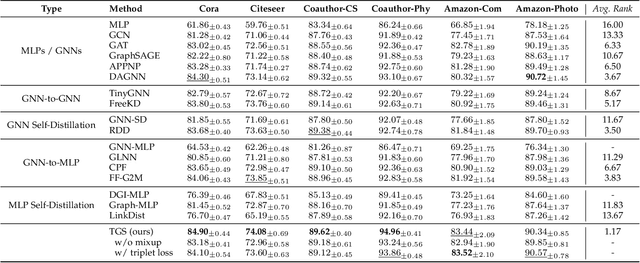
Recent years have witnessed great success in handling graph-related tasks with Graph Neural Networks (GNNs). Despite their great academic success, Multi-Layer Perceptrons (MLPs) remain the primary workhorse for practical industrial applications. One reason for such an academic-industry gap is the neighborhood-fetching latency incurred by data dependency in GNNs. To reduce their gaps, Graph Knowledge Distillation (GKD) is proposed, usually based on a standard teacher-student architecture, to distill knowledge from a large teacher GNN into a lightweight student GNN or MLP. However, we found in this paper that neither teachers nor GNNs are necessary for graph knowledge distillation. We propose a Teacher-Free Graph Self-Distillation (TGS) framework that does not require any teacher model or GNNs during both training and inference. More importantly, the proposed TGS framework is purely based on MLPs, where structural information is only implicitly used to guide dual knowledge self-distillation between the target node and its neighborhood. As a result, TGS enjoys the benefits of graph topology awareness in training but is free from data dependency in inference. Extensive experiments have shown that the performance of vanilla MLPs can be greatly improved with dual self-distillation, e.g., TGS improves over vanilla MLPs by 15.54% on average and outperforms state-of-the-art GKD algorithms on six real-world datasets. In terms of inference speed, TGS infers 75X-89X faster than existing GNNs and 16X-25X faster than classical inference acceleration methods.
Decoupling Weighing and Selecting for Integrating Multiple Graph Pre-training Tasks
Mar 03, 2024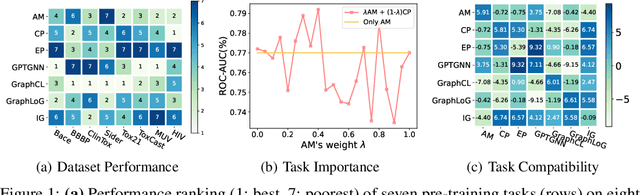
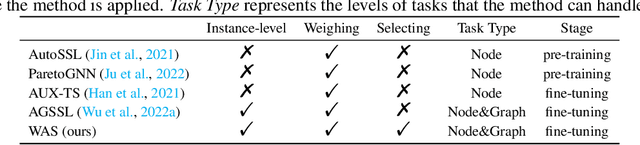
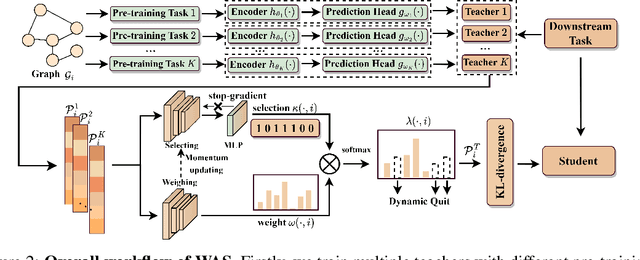
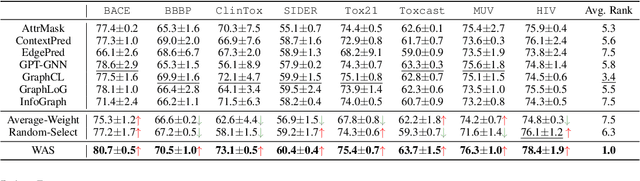
Recent years have witnessed the great success of graph pre-training for graph representation learning. With hundreds of graph pre-training tasks proposed, integrating knowledge acquired from multiple pre-training tasks has become a popular research topic. In this paper, we identify two important collaborative processes for this topic: (1) select: how to select an optimal task combination from a given task pool based on their compatibility, and (2) weigh: how to weigh the selected tasks based on their importance. While there currently has been a lot of work focused on weighing, comparatively little effort has been devoted to selecting. This paper proposes a novel instance-level framework for integrating multiple graph pre-training tasks, Weigh And Select (WAS), where the two collaborative processes, weighing and selecting, are combined by decoupled siamese networks. Specifically, it first adaptively learns an optimal combination of tasks for each instance from a given task pool, based on which a customized instance-level task weighing strategy is learned. Extensive experiments on 16 graph datasets across node-level and graph-level downstream tasks have demonstrated that by combining a few simple but classical tasks, WAS can achieve comparable performance to other leading counterparts. The code is available at https://github.com/TianyuFan0504/WAS.
Enhancing Protein Predictive Models via Proteins Data Augmentation: A Benchmark and New Directions
Mar 01, 2024
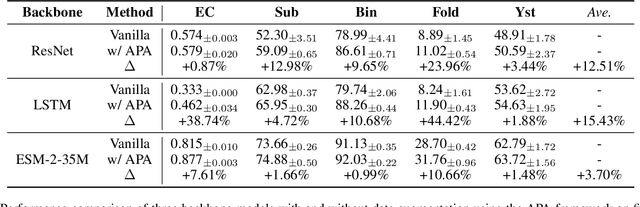
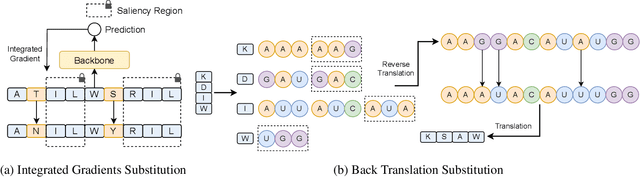
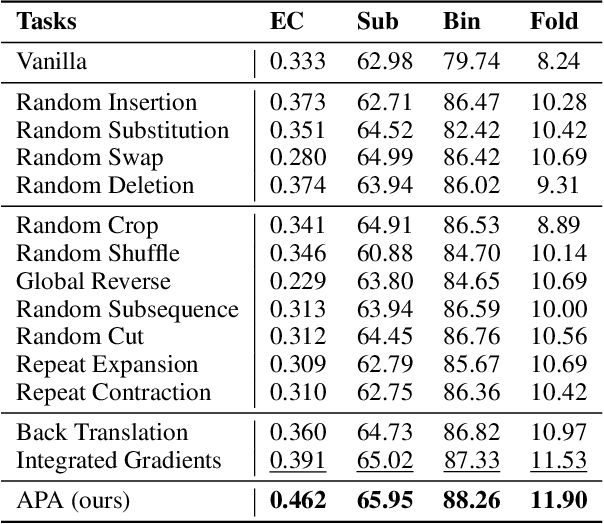
Augmentation is an effective alternative to utilize the small amount of labeled protein data. However, most of the existing work focuses on design-ing new architectures or pre-training tasks, and relatively little work has studied data augmentation for proteins. This paper extends data augmentation techniques previously used for images and texts to proteins and then benchmarks these techniques on a variety of protein-related tasks, providing the first comprehensive evaluation of protein augmentation. Furthermore, we propose two novel semantic-level protein augmentation methods, namely Integrated Gradients Substitution and Back Translation Substitution, which enable protein semantic-aware augmentation through saliency detection and biological knowledge. Finally, we integrate extended and proposed augmentations into an augmentation pool and propose a simple but effective framework, namely Automated Protein Augmentation (APA), which can adaptively select the most suitable augmentation combinations for different tasks. Extensive experiments have shown that APA enhances the performance of five protein related tasks by an average of 10.55% across three architectures compared to vanilla implementations without augmentation, highlighting its potential to make a great impact on the field.
FGBERT: Function-Driven Pre-trained Gene Language Model for Metagenomics
Feb 24, 2024Metagenomic data, comprising mixed multi-species genomes, are prevalent in diverse environments like oceans and soils, significantly impacting human health and ecological functions. However, current research relies on K-mer representations, limiting the capture of structurally relevant gene contexts. To address these limitations and further our understanding of complex relationships between metagenomic sequences and their functions, we introduce a protein-based gene representation as a context-aware and structure-relevant tokenizer. Our approach includes Masked Gene Modeling (MGM) for gene group-level pre-training, providing insights into inter-gene contextual information, and Triple Enhanced Metagenomic Contrastive Learning (TEM-CL) for gene-level pre-training to model gene sequence-function relationships. MGM and TEM-CL constitute our novel metagenomic language model {\NAME}, pre-trained on 100 million metagenomic sequences. We demonstrate the superiority of our proposed {\NAME} on eight datasets.
MAPE-PPI: Towards Effective and Efficient Protein-Protein Interaction Prediction via Microenvironment-Aware Protein Embedding
Feb 22, 2024Protein-Protein Interactions (PPIs) are fundamental in various biological processes and play a key role in life activities. The growing demand and cost of experimental PPI assays require computational methods for efficient PPI prediction. While existing methods rely heavily on protein sequence for PPI prediction, it is the protein structure that is the key to determine the interactions. To take both protein modalities into account, we define the microenvironment of an amino acid residue by its sequence and structural contexts, which describe the surrounding chemical properties and geometric features. In addition, microenvironments defined in previous work are largely based on experimentally assayed physicochemical properties, for which the "vocabulary" is usually extremely small. This makes it difficult to cover the diversity and complexity of microenvironments. In this paper, we propose Microenvironment-Aware Protein Embedding for PPI prediction (MPAE-PPI), which encodes microenvironments into chemically meaningful discrete codes via a sufficiently large microenvironment "vocabulary" (i.e., codebook). Moreover, we propose a novel pre-training strategy, namely Masked Codebook Modeling (MCM), to capture the dependencies between different microenvironments by randomly masking the codebook and reconstructing the input. With the learned microenvironment codebook, we can reuse it as an off-the-shelf tool to efficiently and effectively encode proteins of different sizes and functions for large-scale PPI prediction. Extensive experiments show that MAPE-PPI can scale to PPI prediction with millions of PPIs with superior trade-offs between effectiveness and computational efficiency than the state-of-the-art competitors.
Switch EMA: A Free Lunch for Better Flatness and Sharpness
Feb 14, 2024Exponential Moving Average (EMA) is a widely used weight averaging (WA) regularization to learn flat optima for better generalizations without extra cost in deep neural network (DNN) optimization. Despite achieving better flatness, existing WA methods might fall into worse final performances or require extra test-time computations. This work unveils the full potential of EMA with a single line of modification, i.e., switching the EMA parameters to the original model after each epoch, dubbed as Switch EMA (SEMA). From both theoretical and empirical aspects, we demonstrate that SEMA can help DNNs to reach generalization optima that better trade-off between flatness and sharpness. To verify the effectiveness of SEMA, we conduct comparison experiments with discriminative, generative, and regression tasks on vision and language datasets, including image classification, self-supervised learning, object detection and segmentation, image generation, video prediction, attribute regression, and language modeling. Comprehensive results with popular optimizers and networks show that SEMA is a free lunch for DNN training by improving performances and boosting convergence speeds.
PSC-CPI: Multi-Scale Protein Sequence-Structure Contrasting for Efficient and Generalizable Compound-Protein Interaction Prediction
Feb 13, 2024Compound-Protein Interaction (CPI) prediction aims to predict the pattern and strength of compound-protein interactions for rational drug discovery. Existing deep learning-based methods utilize only the single modality of protein sequences or structures and lack the co-modeling of the joint distribution of the two modalities, which may lead to significant performance drops in complex real-world scenarios due to various factors, e.g., modality missing and domain shifting. More importantly, these methods only model protein sequences and structures at a single fixed scale, neglecting more fine-grained multi-scale information, such as those embedded in key protein fragments. In this paper, we propose a novel multi-scale Protein Sequence-structure Contrasting framework for CPI prediction (PSC-CPI), which captures the dependencies between protein sequences and structures through both intra-modality and cross-modality contrasting. We further apply length-variable protein augmentation to allow contrasting to be performed at different scales, from the amino acid level to the sequence level. Finally, in order to more fairly evaluate the model generalizability, we split the test data into four settings based on whether compounds and proteins have been observed during the training stage. Extensive experiments have shown that PSC-CPI generalizes well in all four settings, particularly in the more challenging ``Unseen-Both" setting, where neither compounds nor proteins have been observed during training. Furthermore, even when encountering a situation of modality missing, i.e., inference with only single-modality protein data, PSC-CPI still exhibits comparable or even better performance than previous approaches.
A Graph is Worth $K$ Words: Euclideanizing Graph using Pure Transformer
Feb 04, 2024Can we model non-Euclidean graphs as pure language or even Euclidean vectors while retaining their inherent information? The non-Euclidean property have posed a long term challenge in graph modeling. Despite recent GNN and Graphformer efforts encoding graphs as Euclidean vectors, recovering original graph from the vectors remains a challenge. We introduce GraphsGPT, featuring a Graph2Seq encoder that transforms non-Euclidean graphs into learnable graph words in a Euclidean space, along with a GraphGPT decoder that reconstructs the original graph from graph words to ensure information equivalence. We pretrain GraphsGPT on 100M molecules and yield some interesting findings: (1) Pretrained Graph2Seq excels in graph representation learning, achieving state-of-the-art results on 8/9 graph classification and regression tasks. (2) Pretrained GraphGPT serves as a strong graph generator, demonstrated by its ability to perform both unconditional and conditional graph generation. (3) Graph2Seq+GraphGPT enables effective graph mixup in the Euclidean space, overcoming previously known non-Euclidean challenge. (4) Our proposed novel edge-centric GPT pretraining task is effective in graph fields, underscoring its success in both representation and generation.