 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePaccMann: Prediction of anticancer compound sensitivity with multi-modal attention-based neural networks
Nov 16, 2018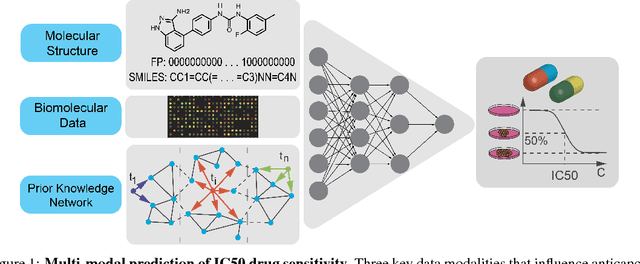
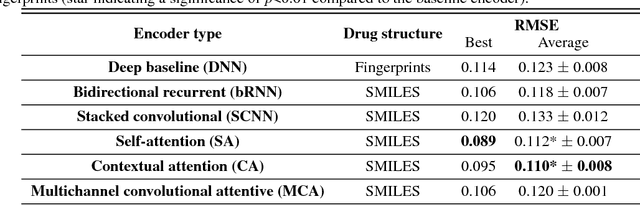
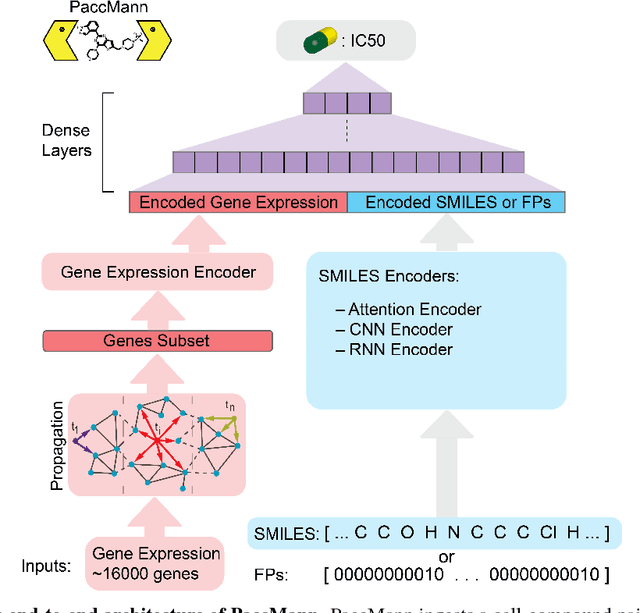
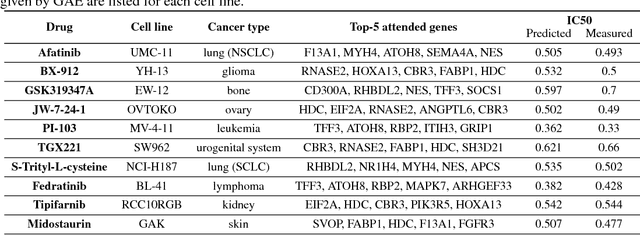
We present a novel approach for the prediction of anticancer compound sensitivity by means of multi-modal attention-based neural networks (PaccMann). In our approach, we integrate three key pillars of drug sensitivity, namely, the molecular structure of compounds, transcriptomic profiles of cancer cells as well as prior knowledge about interactions among proteins within cells. Our models ingest a drug-cell pair consisting of SMILES encoding of a compound and the gene expression profile of a cancer cell and predicts an IC50 sensitivity value. Gene expression profiles are encoded using an attention-based encoding mechanism that assigns high weights to the most informative genes. We present and study three encoders for SMILES string of compounds: 1) bidirectional recurrent 2) convolutional 3) attention-based encoders. We compare our devised models against a baseline model that ingests engineered fingerprints to represent the molecular structure. We demonstrate that using our attention-based encoders, we can surpass the baseline model. The use of attention-based encoders enhance interpretability and enable us to identify genes, bonds and atoms that were used by the network to make a prediction.
Network-based Biased Tree Ensembles for Drug Sensitivity Prediction and Drug Sensitivity Biomarker Identification in Cancer
Aug 18, 2018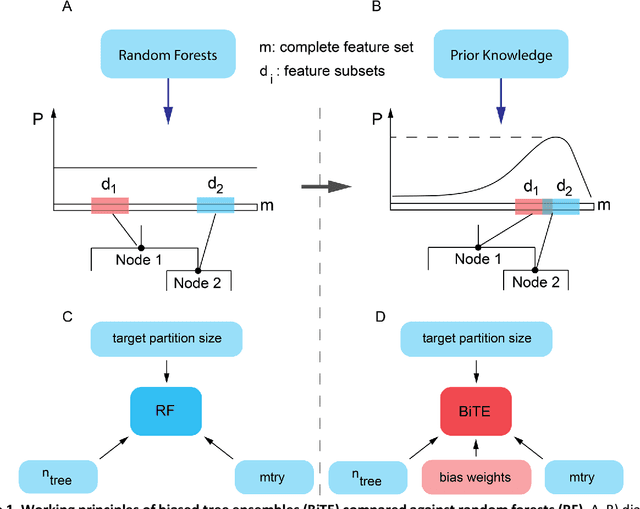
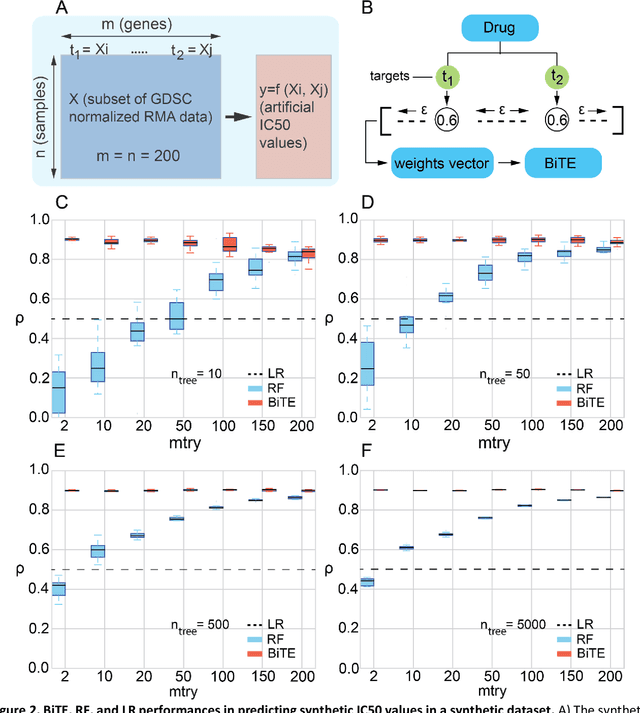
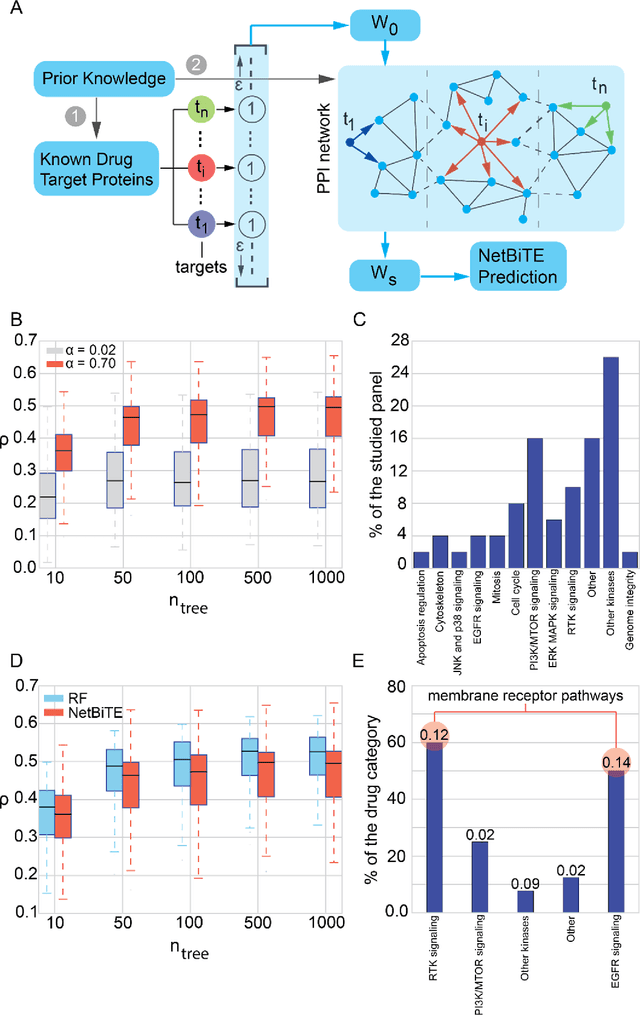
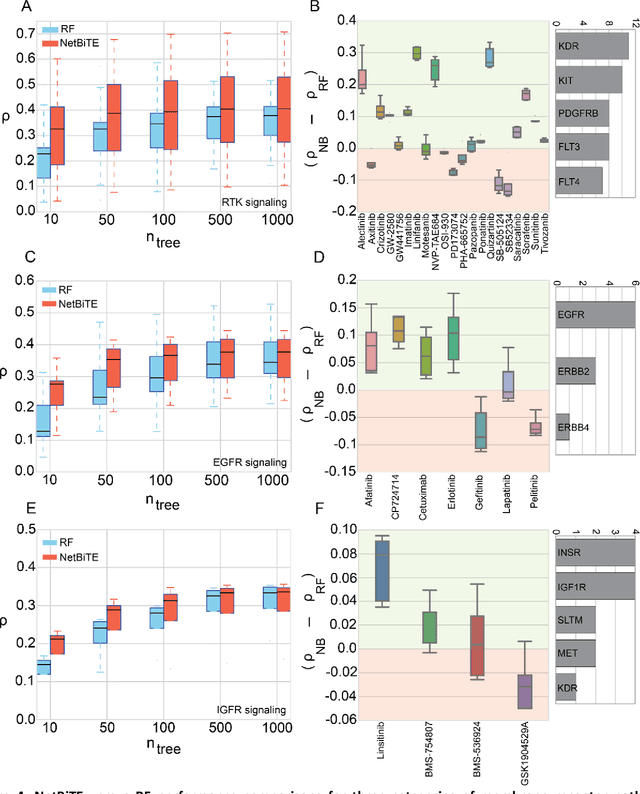
We present the Network-based Biased Tree Ensembles (NetBiTE) method for drug sensitivity prediction and drug sensitivity biomarker identification in cancer using a combination of prior knowledge and gene expression data. Our devised method consists of a biased tree ensemble that is built according to a probabilistic bias weight distribution. The bias weight distribution is obtained from the assignment of high weights to the drug targets and propagating the assigned weights over a protein-protein interaction network such as STRING. The propagation of weights, defines neighborhoods of influence around the drug targets and as such simulates the spread of perturbations within the cell, following drug administration. Using a synthetic dataset, we showcase how application of biased tree ensembles (BiTE) results in significant accuracy gains at a much lower computational cost compared to the unbiased random forests (RF) algorithm. We then apply NetBiTE to the Genomics of Drug Sensitivity in Cancer (GDSC) dataset and demonstrate that NetBiTE outperforms RF in predicting IC50 drug sensitivity, only for drugs that target membrane receptor pathways (MRPs): RTK, EGFR and IGFR signaling pathways. We propose based on the NetBiTE results, that for drugs that inhibit MRPs, the expression of target genes prior to drug administration is a biomarker for IC50 drug sensitivity following drug administration. We further verify and reinforce this proposition through control studies on, PI3K/MTOR signaling pathway inhibitors, a drug category that does not target MRPs, and through assignment of dummy targets to MRP inhibiting drugs and investigating the variation in NetBiTE accuracy.
PIMKL: Pathway Induced Multiple Kernel Learning
Jul 05, 2018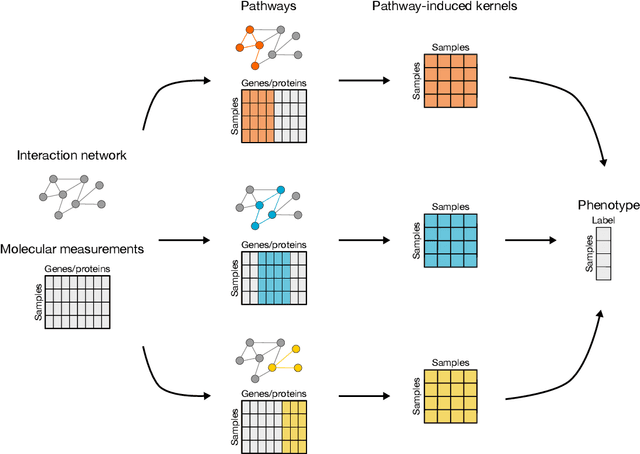
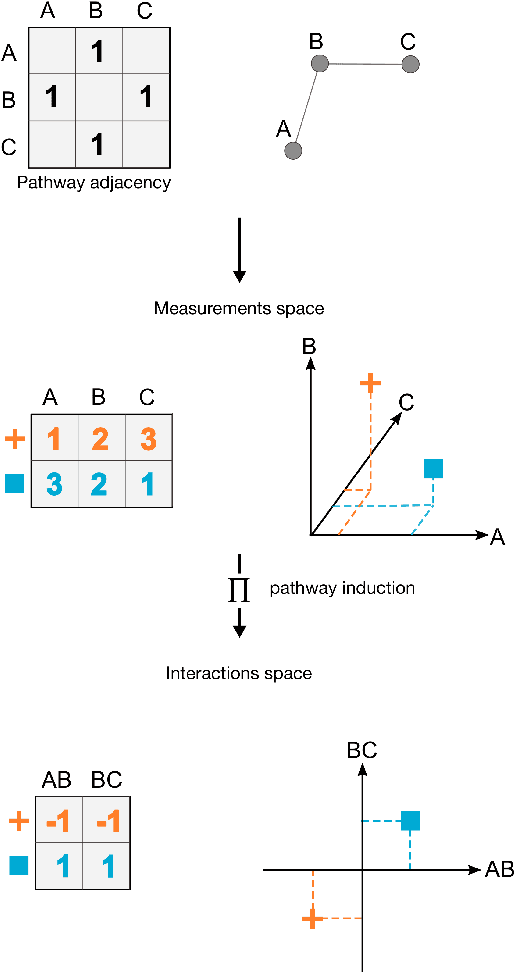
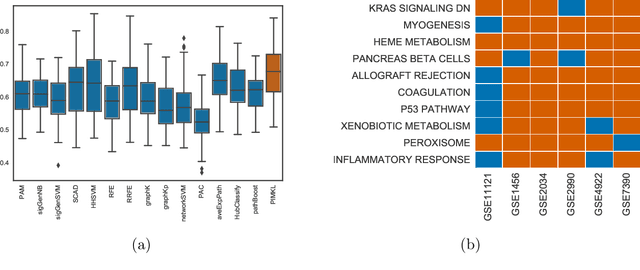
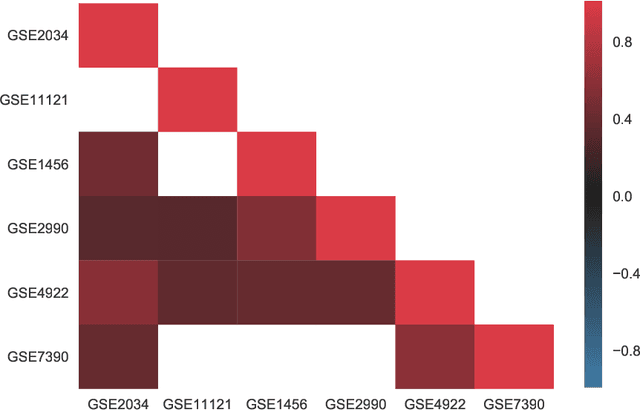
Reliable identification of molecular biomarkers is essential for accurate patient stratification. While state-of-the-art machine learning approaches for sample classification continue to push boundaries in terms of performance, most of these methods are not able to integrate different data types and lack generalization power, limiting their application in a clinical setting. Furthermore, many methods behave as black boxes, and we have very little understanding about the mechanisms that lead to the prediction. While opaqueness concerning machine behaviour might not be a problem in deterministic domains, in health care, providing explanations about the molecular factors and phenotypes that are driving the classification is crucial to build trust in the performance of the predictive system. We propose Pathway Induced Multiple Kernel Learning (PIMKL), a novel methodology to reliably classify samples that can also help gain insights into the molecular mechanisms that underlie the classification. PIMKL exploits prior knowledge in the form of a molecular interaction network and annotated gene sets, by optimizing a mixture of pathway-induced kernels using a Multiple Kernel Learning (MKL) algorithm, an approach that has demonstrated excellent performance in different machine learning applications. After optimizing the combination of kernels for prediction of a specific phenotype, the model provides a stable molecular signature that can be interpreted in the light of the ingested prior knowledge and that can be used in transfer learning tasks.