 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDebiasedDTA: Model Debiasing to Boost Drug-Target Affinity Prediction
Jul 17, 2021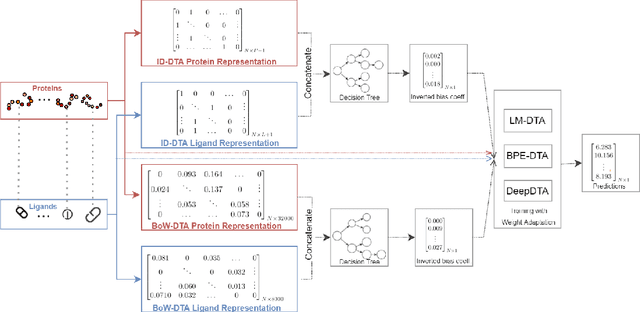
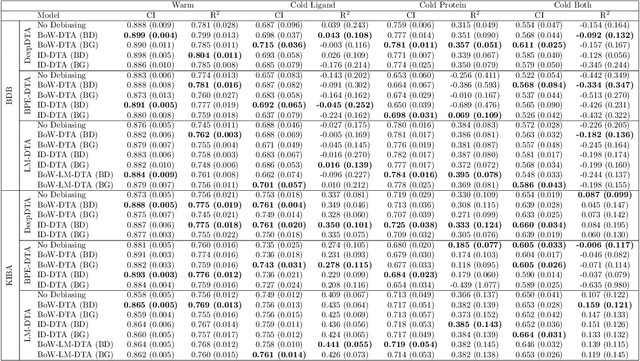
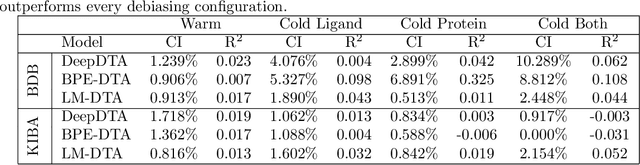
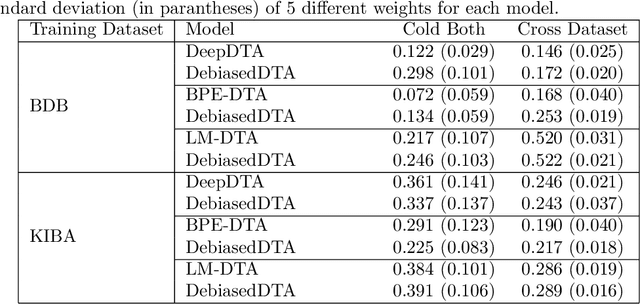
Motivation: Computational models that accurately identify high-affinity protein-compound pairs can accelerate drug discovery pipelines. These models aim to learn binding mechanics through drug-target interaction datasets and use the learned knowledge for predicting the affinity of an input protein-compound pair. However, the datasets they rely on bear misleading patterns that bias models towards memorizing dataset-specific biomolecule properties, instead of learning binding mechanics. This results in models that struggle while predicting drug-target affinities (DTA), especially between de novo biomolecules. Here we present DebiasedDTA, the first DTA model debiasing approach that avoids dataset biases in order to boost affinity prediction for novel biomolecules. DebiasedDTA uses ensemble learning and sample weight adaptation for bias identification and avoidance and is applicable to almost all existing DTA prediction models. Results: The results show that DebiasedDTA can boost models while predicting the interactions between novel biomolecules. Known biomolecules also benefit from the performance improvement, especially when the test biomolecules are dissimilar to the training set. The experiments also show that DebiasedDTA can augment DTA prediction models of different input and model structures and is able to avoid biases of different sources. Availability and Implementation: The source code, the models, and the datasets are freely available for download at https://github.com/boun-tabi/debiaseddta-reproduce, implementation in Python3, and supported for Linux, MacOS and MS Windows. Contact: arzucan.ozgur@boun.edu.tr, elif.ozkirimli@roche.com
The RELX Dataset and Matching the Multilingual Blanks for Cross-Lingual Relation Classification
Oct 19, 2020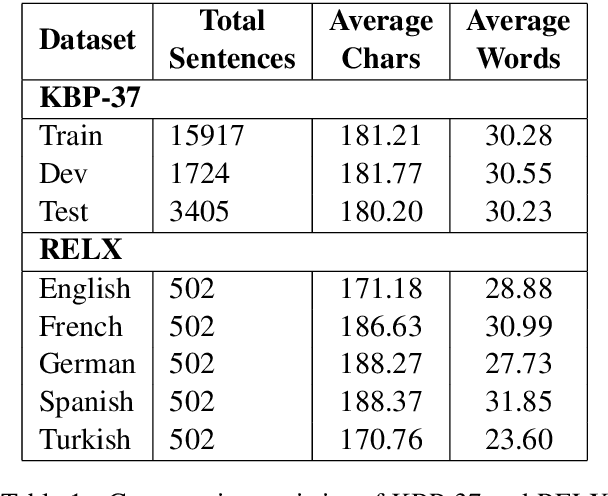
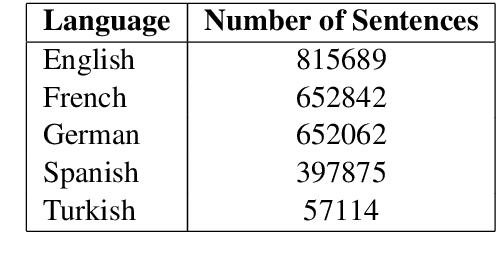
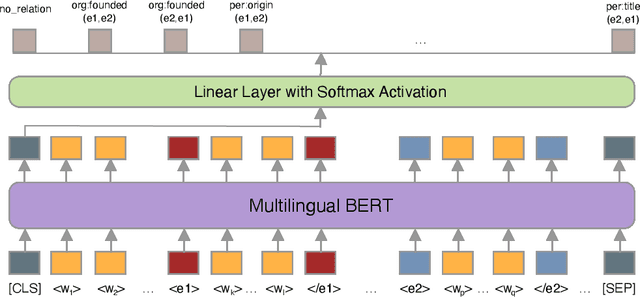

Relation classification is one of the key topics in information extraction, which can be used to construct knowledge bases or to provide useful information for question answering. Current approaches for relation classification are mainly focused on the English language and require lots of training data with human annotations. Creating and annotating a large amount of training data for low-resource languages is impractical and expensive. To overcome this issue, we propose two cross-lingual relation classification models: a baseline model based on Multilingual BERT and a new multilingual pretraining setup, which significantly improves the baseline with distant supervision. For evaluation, we introduce a new public benchmark dataset for cross-lingual relation classification in English, French, German, Spanish, and Turkish, called RELX. We also provide the RELX-Distant dataset, which includes hundreds of thousands of sentences with relations from Wikipedia and Wikidata collected by distant supervision for these languages. Our code and data are available at: https://github.com/boun-tabi/RELX
Vapur: A Search Engine to Find Related Protein -- Compound Pairs in COVID-19 Literature
Sep 05, 2020Coronavirus Disease of 2019 (COVID-19) created dire consequences globally and triggered an enormous scientific effort from different domains. Resulting publications formed a gigantic domain-specific collection of text in which finding studies on a biomolecule of interest is quite challenging for general purpose search engines due to terminology-rich characteristics of the publications. Here, we present Vapur, an online COVID-19 search engine specifically designed for finding related protein - chemical pairs. Vapur is empowered with a biochemically related entities-oriented inverted index in order to group studies relevant to a biomolecule with respect to its related entities. The inverted index of Vapur is automatically created with a BioNLP pipeline and integrated with an online user interface. The online interface is designed for the smooth traversal of the current literature and is publicly available at https://tabilab.cmpe.boun.edu.tr/vapur/.
Resources for Turkish Dependency Parsing: Introducing the BOUN Treebank and the BoAT Annotation Tool
Feb 24, 2020

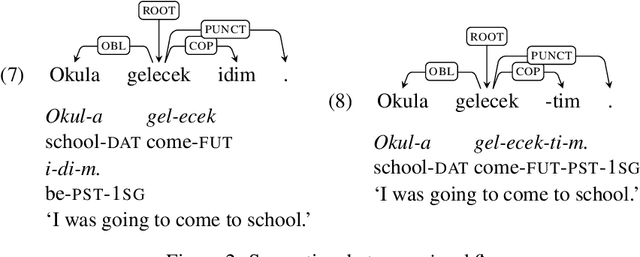
In this paper, we describe our contributions and efforts to develop Turkish resources, which include a new treebank (BOUN Treebank) with novel sentences, along with the guidelines we adopted and a new annotation tool we developed (BoAT). The manual annotation process we employed was shaped and implemented by a team of four linguists and five NLP specialists. Decisions regarding the annotation of the BOUN Treebank were made in line with the Universal Dependencies framework, which originated from the works of De Marneffe et al. (2014) and Nivre et al. (2016). We took into account the recent unifying efforts based on the re-annotation of other Turkish treebanks in the UD framework (T\"urk et al., 2019). Through the BOUN Treebank, we introduced a total of 9,757 sentences from various topics including biographical texts, national newspapers, instructional texts, popular culture articles, and essays. In addition, we report the parsing results of a graph-based dependency parser obtained over each text type, the total of the BOUN Treebank, and all Turkish treebanks that we either re-annotated or introduced. We show that a state-of-the-art dependency parser has improved scores for identifying the proper head and the syntactic relationships between the heads and the dependents. In light of these results, we have observed that the unification of the Turkish annotation scheme and introducing a more comprehensive treebank improves performance with regards to dependency parsing
A Hybrid Approach to Dependency Parsing: Combining Rules and Morphology with Deep Learning
Feb 24, 2020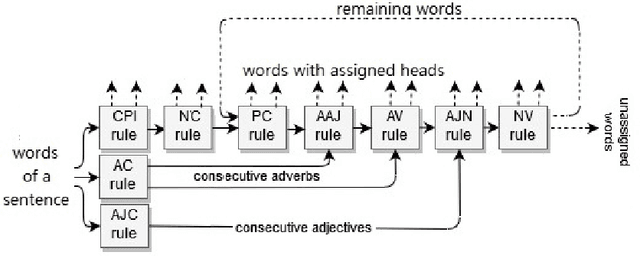



Fully data-driven, deep learning-based models are usually designed as language-independent and have been shown to be successful for many natural language processing tasks. However, when the studied language is low-resourced and the amount of training data is insufficient, these models can benefit from the integration of natural language grammar-based information. We propose two approaches to dependency parsing especially for languages with restricted amount of training data. Our first approach combines a state-of-the-art deep learning-based parser with a rule-based approach and the second one incorporates morphological information into the parser. In the rule-based approach, the parsing decisions made by the rules are encoded and concatenated with the vector representations of the input words as additional information to the deep network. The morphology-based approach proposes different methods to include the morphological structure of words into the parser network. Experiments are conducted on the IMST-UD Treebank and the results suggest that integration of explicit knowledge about the target language to a neural parser through a rule-based parsing system and morphological analysis leads to more accurate annotations and hence, increases the parsing performance in terms of attachment scores. The proposed methods are developed for Turkish, but can be adapted to other languages as well.
Exploring Chemical Space using Natural Language Processing Methodologies for Drug Discovery
Feb 10, 2020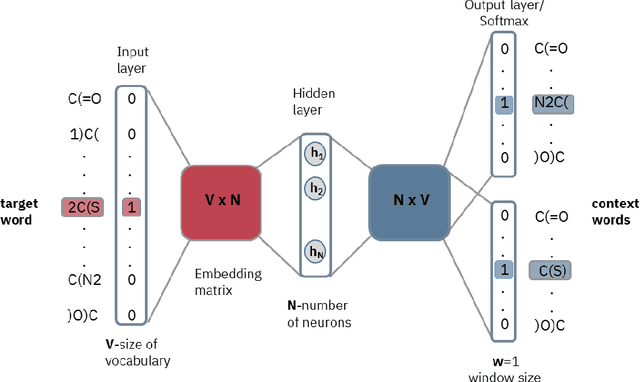
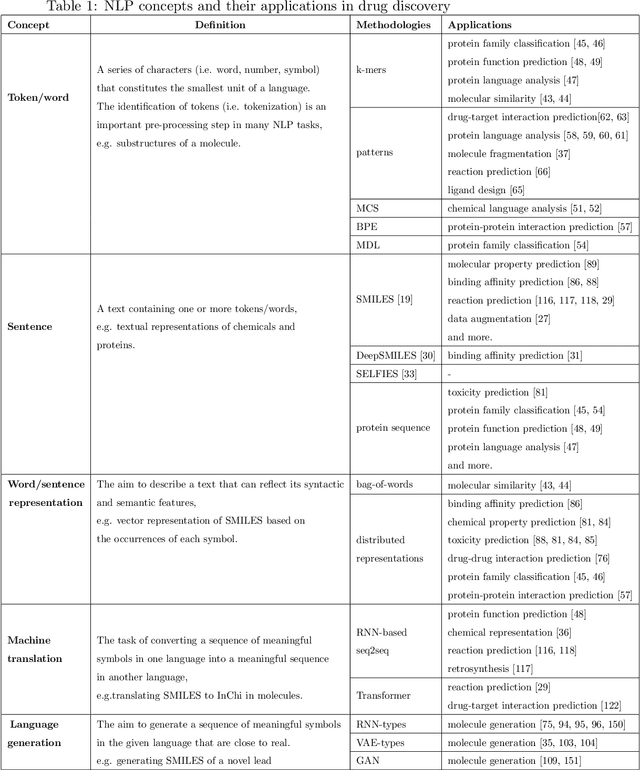
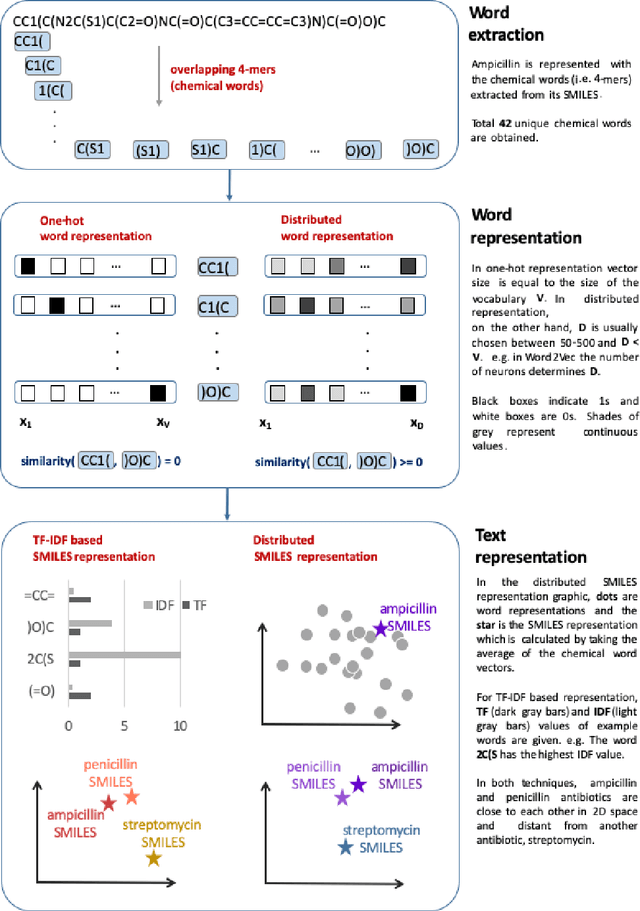
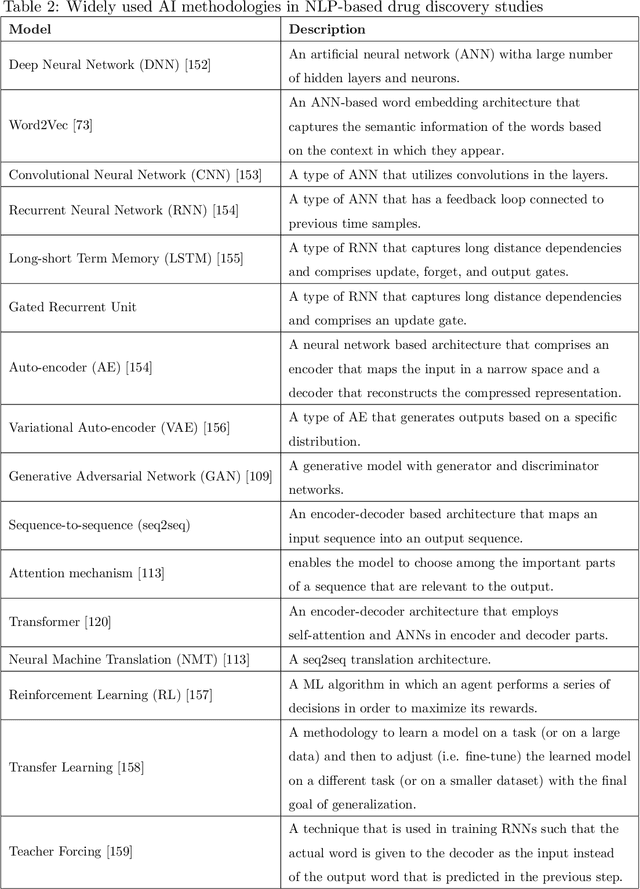
Text-based representations of chemicals and proteins can be thought of as unstructured languages codified by humans to describe domain-specific knowledge. Advances in natural language processing (NLP) methodologies in the processing of spoken languages accelerated the application of NLP to elucidate hidden knowledge in textual representations of these biochemical entities and then use it to construct models to predict molecular properties or to design novel molecules. This review outlines the impact made by these advances on drug discovery and aims to further the dialogue between medicinal chemists and computer scientists.
WideDTA: prediction of drug-target binding affinity
Feb 04, 2019
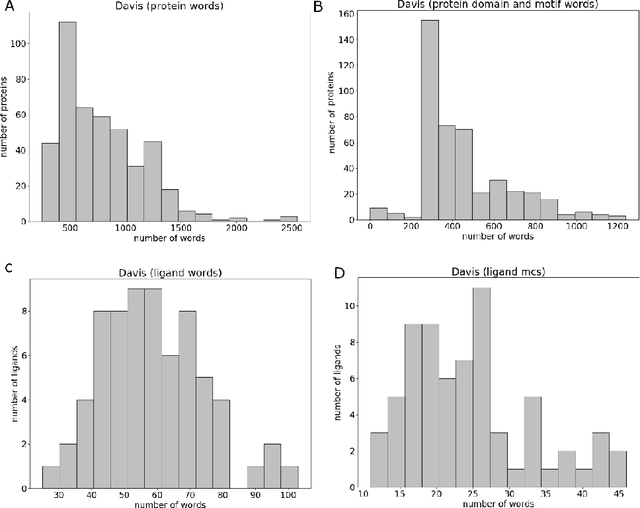
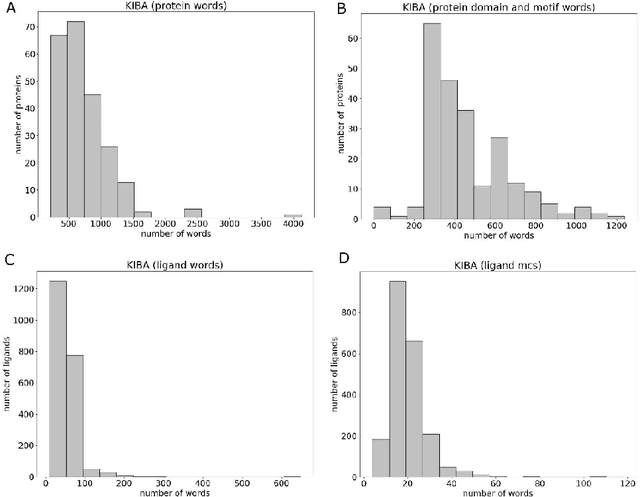
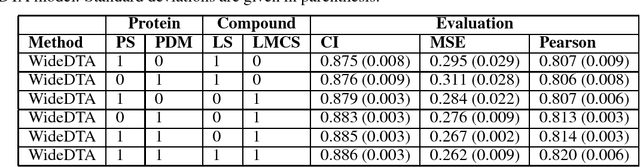
Motivation: Prediction of the interaction affinity between proteins and compounds is a major challenge in the drug discovery process. WideDTA is a deep-learning based prediction model that employs chemical and biological textual sequence information to predict binding affinity. Results: WideDTA uses four text-based information sources, namely the protein sequence, ligand SMILES, protein domains and motifs, and maximum common substructure words to predict binding affinity. WideDTA outperformed one of the state of the art deep learning methods for drug-target binding affinity prediction, DeepDTA on the KIBA dataset with a statistical significance. This indicates that the word-based sequence representation adapted by WideDTA is a promising alternative to the character-based sequence representation approach in deep learning models for binding affinity prediction, such as the one used in DeepDTA. In addition, the results showed that, given the protein sequence and ligand SMILES, the inclusion of protein domain and motif information as well as ligand maximum common substructure words do not provide additional useful information for the deep learning model. Interestingly, however, using only domain and motif information to represent proteins achieved similar performance to using the full protein sequence, suggesting that important binding relevant information is contained within the protein motifs and domains.
A chemical language based approach for protein - ligand interaction prediction
Nov 02, 2018
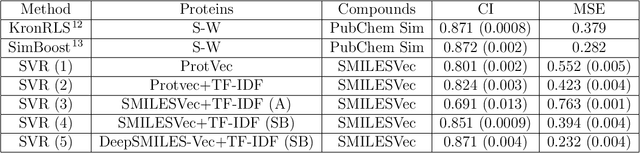
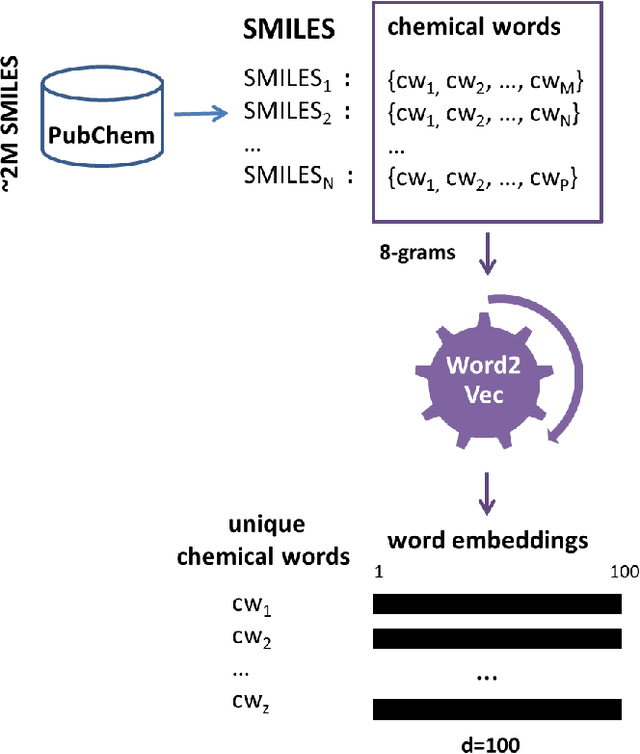
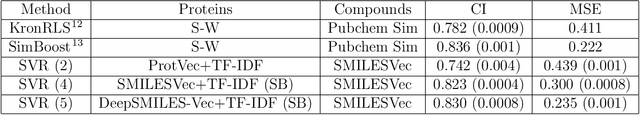
Identification of high affinity drug-target interactions (DTI) is a major research question in drug discovery. In this study, we propose a novel methodology to predict drug-target binding affinity using only ligand SMILES information. We represent proteins using the word-embeddings of the SMILES representations of their strong binding ligands. Each SMILES is represented in the form of a set of chemical words and a protein is described by the set of chemical words with the highest Term Frequency- Inverse Document Frequency (TF-IDF) value. We then utilize the Support Vector Regression (SVR) algorithm to predict protein - drug binding affinities in the Davis and KIBA Kinase datasets. We also compared the performance of SMILES representation with the recently proposed DeepSMILES representation and found that using DeepSMILES yields better performance in the prediction task. Using only SMILESVec, which is a strictly string based representation of the proteins based on their interacting ligands, we were able to predict drug-target binding affinity as well as or better than the KronRLS or SimBoost models that utilize protein sequence.
Named Entity Recognition on Twitter for Turkish using Semi-supervised Learning with Word Embeddings
Oct 20, 2018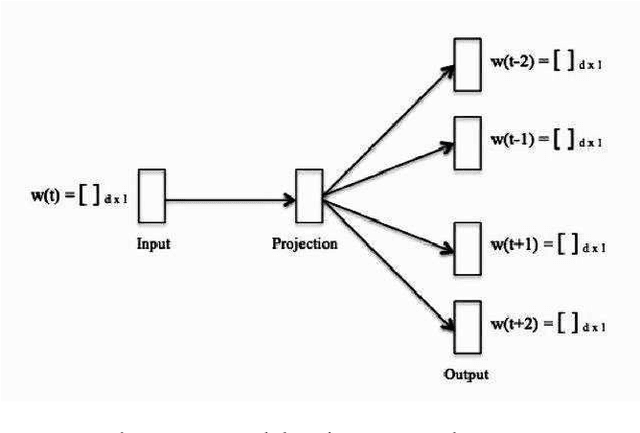
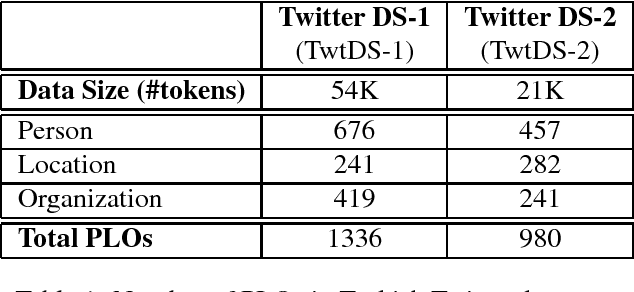
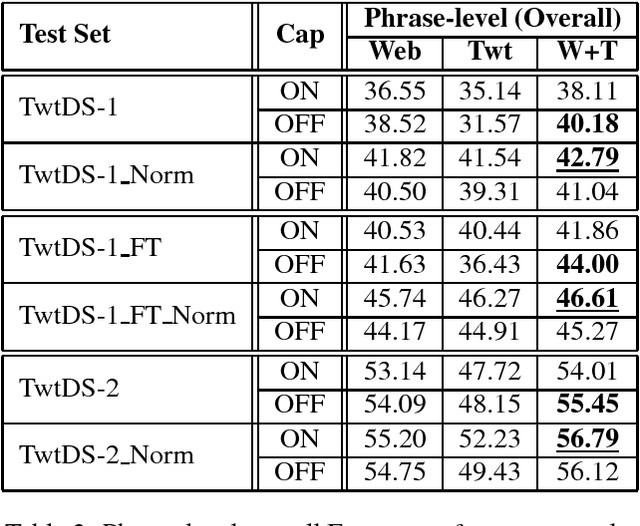
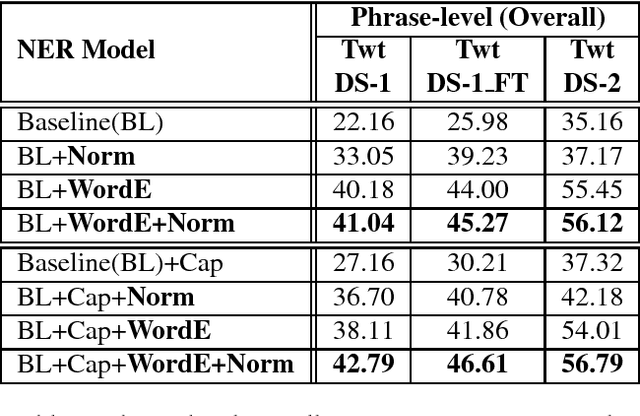
Recently, due to the increasing popularity of social media, the necessity for extracting information from informal text types, such as microblog texts, has gained significant attention. In this study, we focused on the Named Entity Recognition (NER) problem on informal text types for Turkish. We utilized a semi-supervised learning approach based on neural networks. We applied a fast unsupervised method for learning continuous representations of words in vector space. We made use of these obtained word embeddings, together with language independent features that are engineered to work better on informal text types, for generating a Turkish NER system on microblog texts. We evaluated our Turkish NER system on Twitter messages and achieved better F-score performances than the published results of previously proposed NER systems on Turkish tweets. Since we did not employ any language dependent features, we believe that our method can be easily adapted to microblog texts in other morphologically rich languages.
DeepDTA: Deep Drug-Target Binding Affinity Prediction
Jun 05, 2018
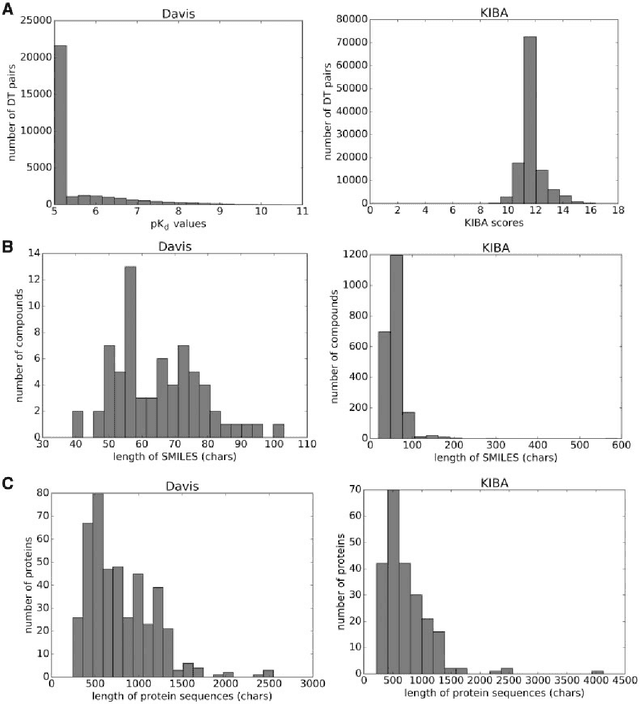
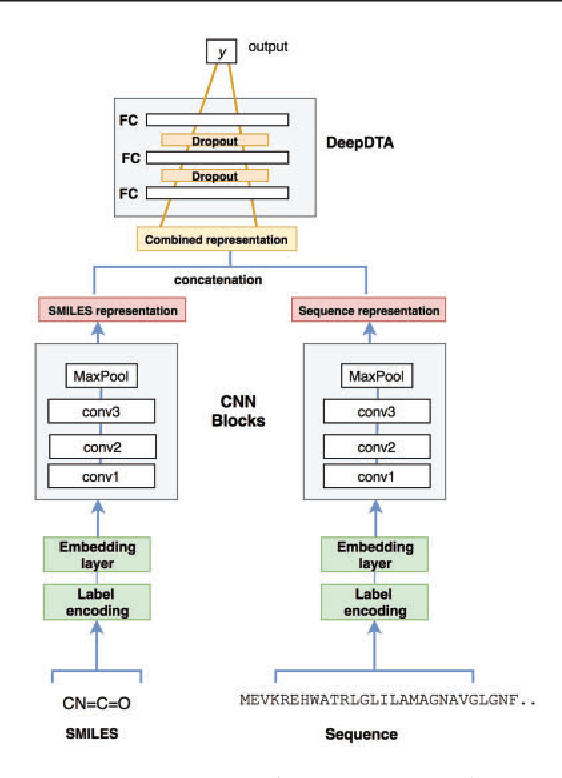

The identification of novel drug-target (DT) interactions is a substantial part of the drug discovery process. Most of the computational methods that have been proposed to predict DT interactions have focused on binary classification, where the goal is to determine whether a DT pair interacts or not. However, protein-ligand interactions assume a continuum of binding strength values, also called binding affinity and predicting this value still remains a challenge. The increase in the affinity data available in DT knowledge-bases allows the use of advanced learning techniques such as deep learning architectures in the prediction of binding affinities. In this study, we propose a deep-learning based model that uses only sequence information of both targets and drugs to predict DT interaction binding affinities. The few studies that focus on DT binding affinity prediction use either 3D structures of protein-ligand complexes or 2D features of compounds. One novel approach used in this work is the modeling of protein sequences and compound 1D representations with convolutional neural networks (CNNs). The results show that the proposed deep learning based model that uses the 1D representations of targets and drugs is an effective approach for drug target binding affinity prediction. The model in which high-level representations of a drug and a target are constructed via CNNs achieved the best Concordance Index (CI) performance in one of our larger benchmark data sets, outperforming the KronRLS algorithm and SimBoost, a state-of-the-art method for DT binding affinity prediction.